×
Close
Sign Up
Login
Home
SCI Abstract
Library D
Events & Partner
WeMed
MDLA Events Platform
Events
Media Partners
Educational Partner
User Tools
FAQ/USER GUIDE
Language
English
中文/ Chinese
French
Português
Español
Arabic
Russian
Contact Us
×
Close
mdla_1
mdla_2
mdla_3
mdla_4
mdla_5
mdla_6
Categories
Biochemistry & Molecular Biology
30077
Global Medical University
5141
Allergy
1792
Anatomy & Morphology
1584
Andrology
379
Anesthesia & Intensive Care
1300
Anesthesiology
5399
Audiology & Speech-Language Pathology
347
Behavioral Sciences
105
Biochemical Research Methods
7243
Biodiversity Conservation
330
Biology
8515
Biophysics
8292
Biotechnology & Applied Microbiology
8662
Cardiac & Cardiovascular Systems
31125
Cardiovascular & Respiratory Systems
1413
Cell & Tissue Engineering
704
Cell Biology
11182
Chemistry, Analytical
4301
Chemistry, Applied
10948
Chemistry, Medicinal
8755
Chemistry, Multidisciplinary
18521
Clinical Immunology & Infectious Disease
423
Clinical Medicine
8962
Clinical Neurology
16636
Clinical Psychology & Psychiatry
1294
Critical Care Medicine
3163
Dentistry, Oral Surgery & Medicine
13544
Dermatology
7485
Developmental Biology
6954
Ecology
633
Education, Scientific Disciplines
2050
Emergency Medicine
4117
Endocrinology, Metabolism & Nutrition
24622
Engineering, Biomedical
3729
Entomology
489
Environmental Medicine & Public Health
4669
Evolutionary Biology
276
Gastroenterology & Hepatology
12236
General & Internal Medicine
7013
Genetics & Heredity
15196
Geriatrics & Gerontology
5075
Gerontology
382
Health Care Sciences & Services
16165
Health Policy & Services
604
Hematology
5578
Immunology
24850
Infectious Diseases
14017
Integrative & Complementary Medicine
2943
Medical Ethics
1239
Medical Informatics
2232
Medical Laboratory Technology
419
Medicine, General & Internal
44379
Medicine, Legal
532
Medicine, Research & Experimental
17792
Microbiology
23387
Mycology
0
Nanoscience & Nanotechnology
5388
Neuroimaging
1365
Neurology
4562
Neurosciences
39861
Nursing
9787
Nutrition & Dietetics
7849
Obstetrics & Gynecology
8277
Oncology
52506
Ophthalmology
9749
Optics
4279
Orthopedics
11803
Orthopedics, Rehabilitation & Sports Medicine
1828
Otolaryngology
1583
Otorhinolaryngology
4894
Parasitology
1167
Pathology
5148
Pediatrics
21618
Peripheral Vascular Disease
4767
Pharmacology & Pharmacy
35500
Pharmacology/Toxicology
12212
Physiology
8922
Polymer Science
542
Primary Health Care
837
Psychiatry
19132
Psychology
5240
Psychology, Applied
111
Psychology, Biological
390
Psychology, Clinical
811
Psychology, Developmental
273
Psychology, Educational
164
Psychology, Experimental
171
Psychology, Mathematical
0
Psychology, Multidisciplinary
1694
Psychology, Psychoanalysis
30
Psychology, Social
131
Public Health & Health Care Science
2225
Public, Environmental & Occupational Health
27291
Quantum Science & Technology
0
Radiology, Nuclear Medicine & Imaging
12500
Radiology, Nuclear Medicine & Medical Imaging
8264
Rehabilitation
3074
Remote Sensing
0
Reproductive Biology
2872
Reproductive Medicine
1212
Research/Laboratory Medicine & Medical Technology
4029
Respiratory System
7421
Rheumatology
6029
Social Sciences, Biomedical
1178
Substance Abuse
2771
Surgery
33948
Toxicology
4445
Transplantation
938
Tropical Medicine
300
Urology & Nephrology
12890
Veterinary Sciences
35
Virology
2551
Zoology
0
Channels
JOURNAL OF COMPUTER-AIDED MOLECULAR DESIGN
205
AMERICAN JOURNAL OF PHYSIOLOGY-ENDOCRINOLOGY AND METABOLISM
209
ANTIOXIDANTS
2594
BIOMOLECULES
2187
BIOORGANIC CHEMISTRY
1693
CELL AND BIOSCIENCE
481
CELLULAR & MOLECULAR BIOLOGY LETTERS
245
CELLULAR AND MOLECULAR LIFE SCIENCES
1172
COLLOIDS AND SURFACES B-BIOINTERFACES
1464
COMPUTATIONAL AND STRUCTURAL BIOTECHNOLOGY JOURNAL
1330
CYTOKINE
703
CYTOKINE & GROWTH FACTOR REVIEWS
202
FREE RADICAL BIOLOGY AND MEDICINE
1026
GENOME RESEARCH
480
INTERNATIONAL JOURNAL OF BIOLOGICAL SCIENCES
40
INTERNATIONAL REVIEW OF CELL AND MOLECULAR BIOLOGY
322
JOURNAL OF BIOLOGICAL CHEMISTRY
2328
JOURNAL OF CELLULAR BIOCHEMISTRY
110
JOURNAL OF GENETICS AND GENOMICS
463
JOURNAL OF INORGANIC BIOCHEMISTRY
725
JOURNAL OF INTEGRATIVE PLANT BIOLOGY
85
JOURNAL OF LIPID RESEARCH
24
JOURNAL OF MOLECULAR BIOLOGY
732
JOURNAL OF PHOTOCHEMISTRY AND PHOTOBIOLOGY B-BIOLOGY
428
MOLECULAR BIOLOGY
292
MOLECULAR CARCINOGENESIS
63
NATURE CHEMICAL BIOLOGY
647
NATURE PROTOCOLS
397
NEUROCHEMISTRY INTERNATIONAL
397
PROGRESS IN LIPID RESEARCH
0
REDOX BIOLOGY
0
SCIENCE SIGNALING
167
SIGNAL TRANSDUCTION AND TARGETED THERAPY
966
BIOCHEMISTRY AND CELL BIOLOGY
132
BIOMEDICAL JOURNAL
0
BIOORGANIC & MEDICINAL CHEMISTRY
1
BIOPHYSICAL CHEMISTRY
0
BIOPOLYMERS
28
BIOTECHNIQUES
367
BMC BIOCHEMISTRY
141
CELL BIOCHEMISTRY AND FUNCTION
38
CHEMBIOCHEM
338
CHEMICAL BIOLOGY & DRUG DESIGN
145
CURRENT ISSUES IN MOLECULAR BIOLOGY
546
FRONTIERS IN MOLECULAR BIOSCIENCES
2702
JOURNAL OF FOOD BIOCHEMISTRY
259
JOURNAL OF MOLECULAR RECOGNITION
35
LIPIDS
23
NEUROCHEMICAL RESEARCH
685
PHYSICAL BIOLOGY
119
PROGRESS IN BIOPHYSICS & MOLECULAR BIOLOGY
6
PROTEIN SCIENCE
152
LIPIDS IN HEALTH AND DISEASE
437
VITAMINS AND HORMONES
277
BIOCHEMISTRY INSIGHTS
8
BIOCHEMISTRY MOSCOW-SUPPLEMENT SERIES B-BIOMEDICAL CHEMISTRY
105
INDIAN JOURNAL OF CLINICAL BIOCHEMISTRY
221
PROTEOMES
63
MOLECULAR BIOLOGY RESEARCH COMMUNICATIONS
87
REVIEWS OF PHYSIOLOGY BIOCHEMISTRY AND PHARMACOLOGY
83
GENES & GENOMICS
434
INTERNATIONAL JOURNAL OF PEPTIDE RESEARCH AND THERAPEUTICS
355
JOURNAL OF BIOMOLECULAR NMR
113
SCI Abstract
search
ALL
RECOMMENDED
+
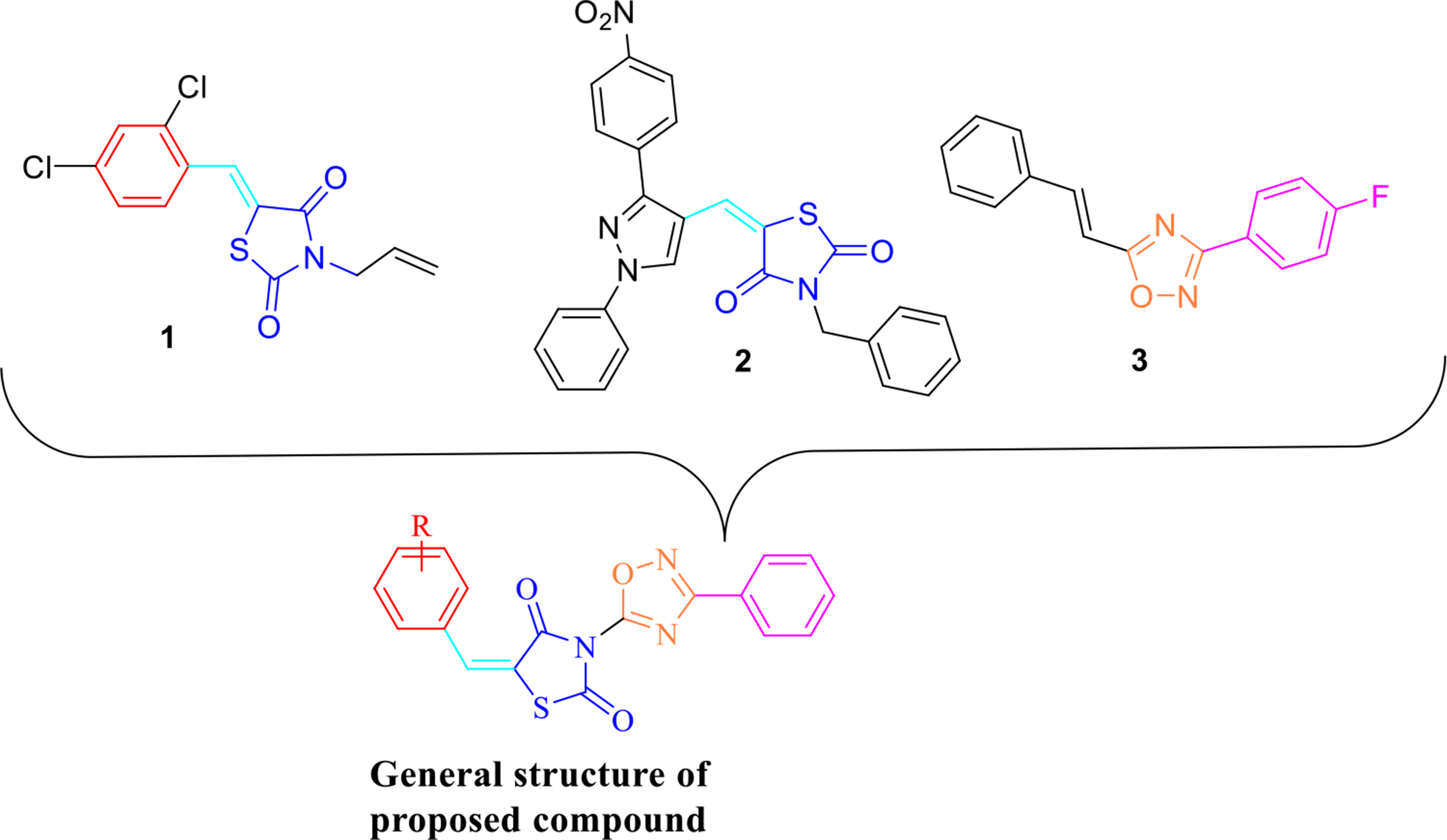
Design and evaluation of novel thiazolidinedione-oxadiazole derivatives as potent α-amylase inhibitors for antidiabetic therapy
Diabetes mellitus is a chronic metabolic disorder characterized by persistent hyperglycemia. Targeting α-amylase, a k...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Protein-ligand co-design: a case for improving binding affinity between type II NADH:quinone oxidoreductase and quinones
Biological engineering aims to enhance biological systems by designing proteins with improved catalytic properties or liga...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Quantum algorithm for protein-ligand docking sites identification in the interaction space
Over the past two decades, the development of novel drugs evolved into a high-demanding computational field. There is a co...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
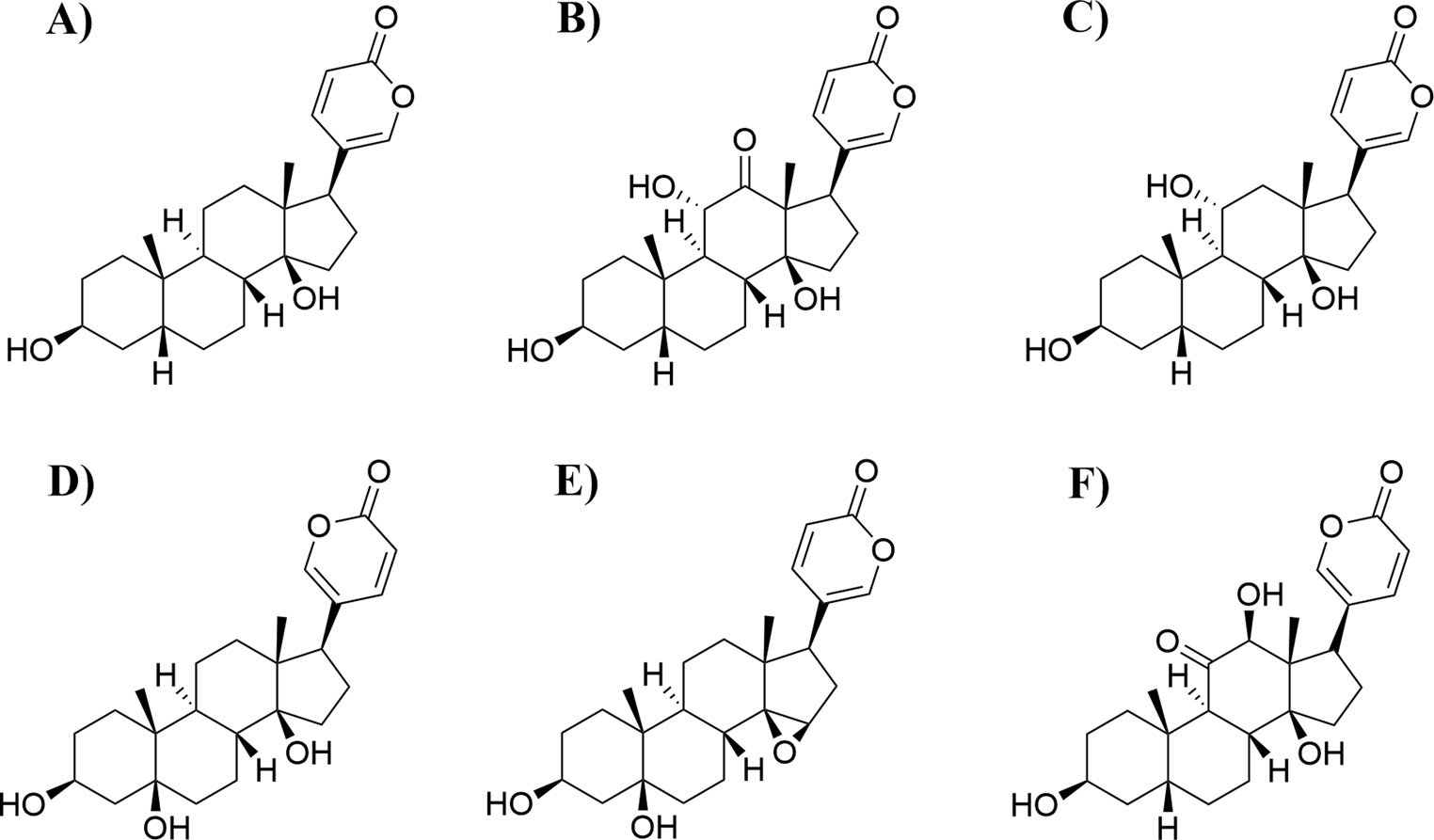
3CL of SARS-CoV-2 as a new target for bufadienolides: in silico and in vitro study
The rapid spread of SARS-CoV-2 and its widespread public health implications have highlighted the urgent need for effectiv...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Unveiling a novel ellagic acid derivative as a potent lipoxygenase (LOX) inhibitor: integration of computational modeling and experimental validation
Cornus macrophylla has been traditionally recognized for its medicinal properties, particularly in managing inflammatory c...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
A computational study of cardiac glycosides from as PI3K inhibitors for targeting HER2 positive breast cancer
The PI3K/Akt pathway plays a crucial role in regulating a broad network of proteins involved in the proliferation of HER2-...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
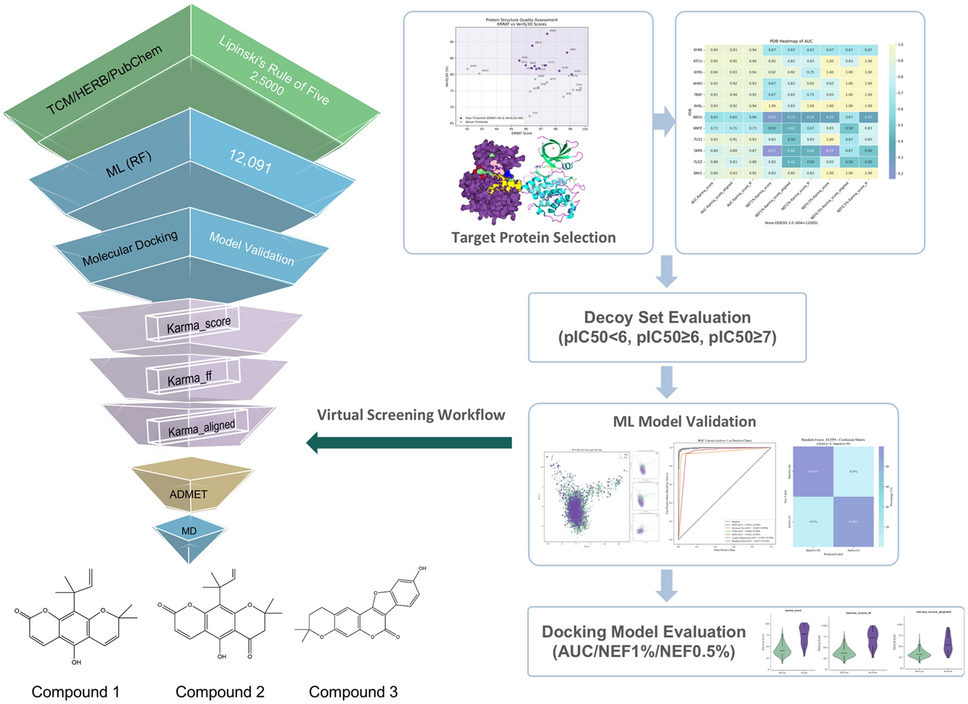
Machine learning-based QSAR and structure-based virtual screening guided discovery of novel mIDH1 inhibitors from natural products
Mutations in isocitrate dehydrogenase 1 (IDH1) have been widely observed in various tumors, such as gliomas and acute myel...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Disrupting tuberculosis pathogenesis by targeting DprE1 in cell wall biosynthesis: a structural dynamics perspective
Mycobacterium tuberculosis (Mtb), the causative agent of TB, remains a major global health challenge due to the emergence ...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
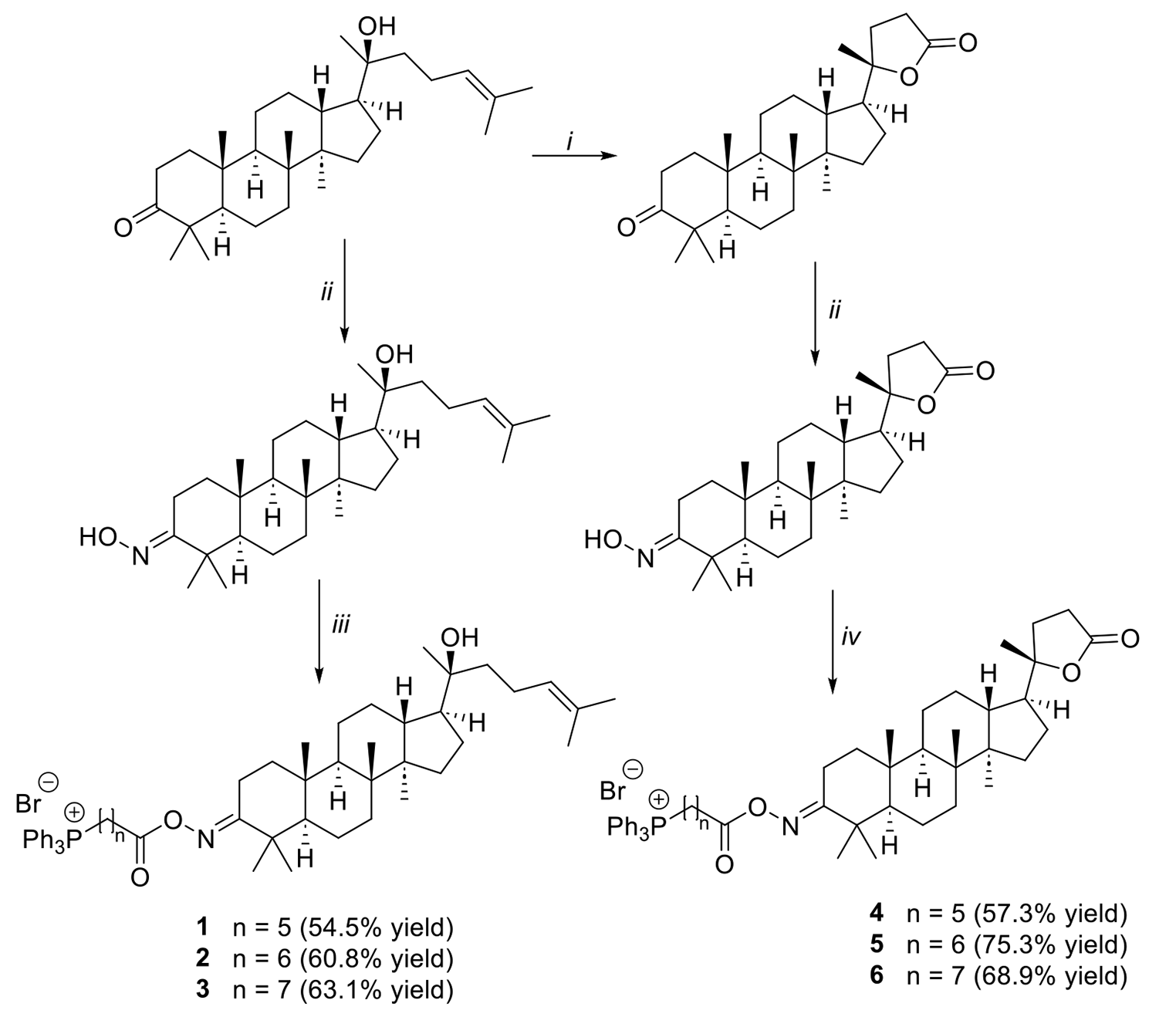
New alkyl triphenylphosphonium dipterocarpol derivatives with cytotoxicity against the MCF-7 breast cancer cell line
Dipterocarpol exhibited cytotoxic properties; however, its hydrophobic nature resulted in decreased bioavailability. This ...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
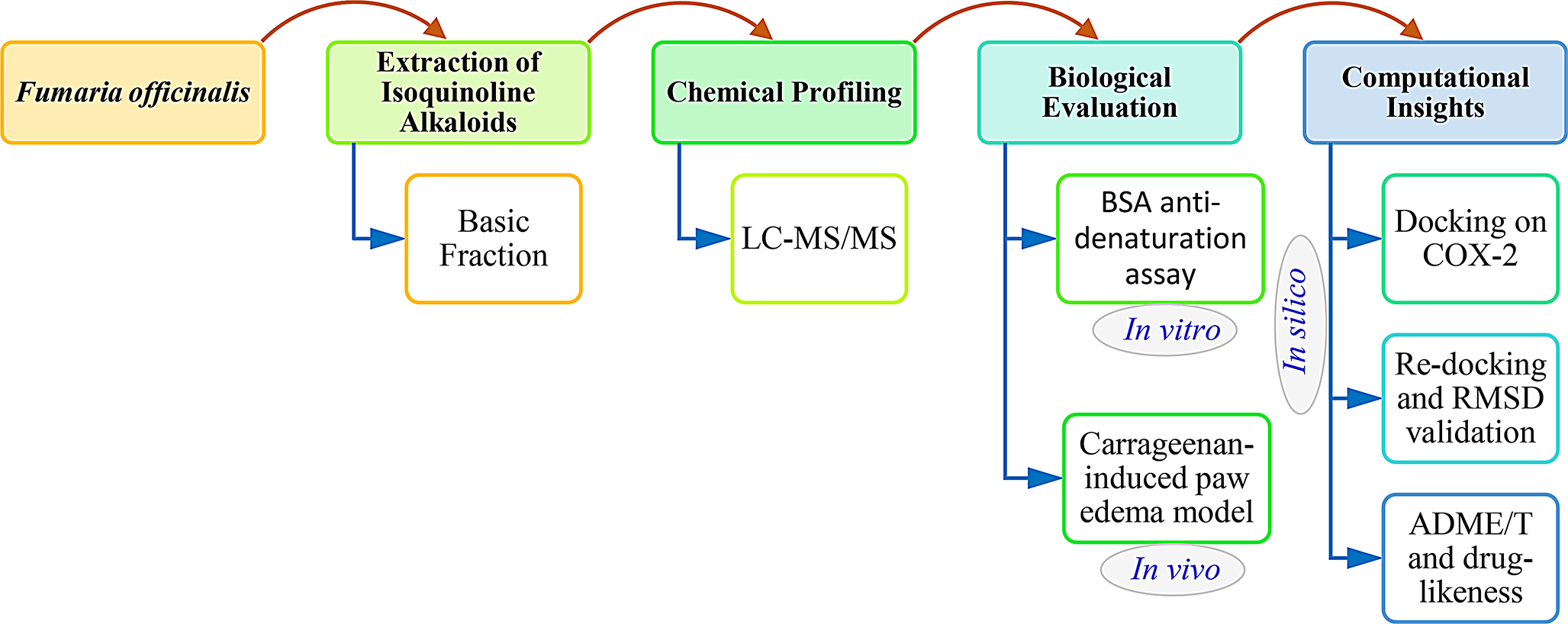
Anti-inflammatory potential of isoquinoline alkaloids from : in vitro, in vivo, and in silico evaluation
The objective of this study is to identify the alkaloids of Fumaria officinalis and to evaluate their anti-inflammatory ac...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
CoBdock-2: enhancing blind docking performance through hybrid feature selection combining ensemble and multimodel feature selection approaches
Identifying orthosteric binding sites and predicting small molecule affinities remains a key challenge in virtual screenin...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
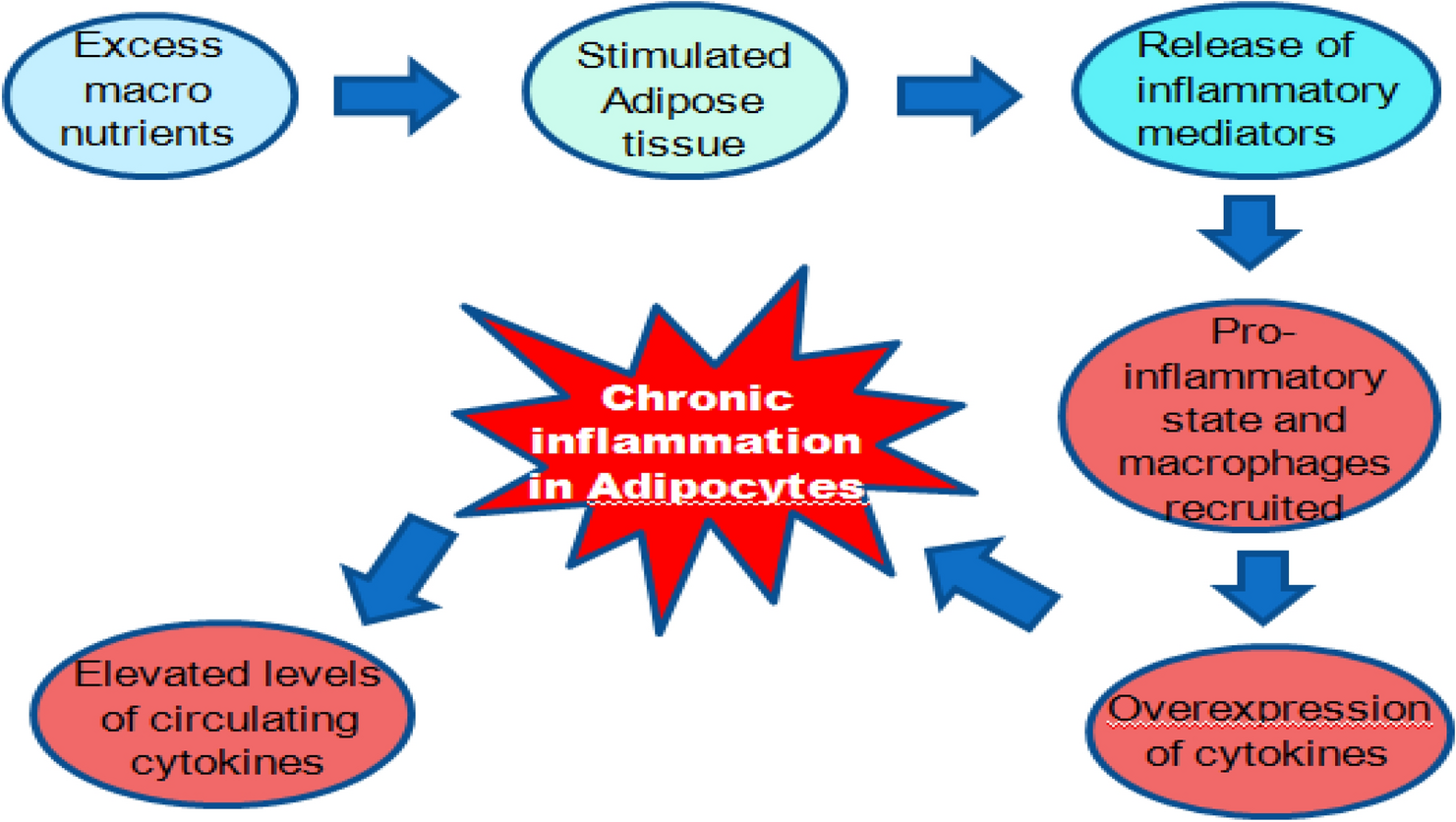
Therapeutic potency of a developed optimized polyherbal formulation in ameliorating obesity induced inflammation and oxidative stress in Swiss albino mice by targeting PPARγ, insulin receptor and AMPK signalling pathway
High fat diet (HFD) induced obesity plays a key role in onset of inflammation, a chronic response of the body to elevated ...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
“Heptadecanol” a phytochemical multi-target inhibitor of SMYD3 & GFPT2 proteins in non-small cell lung cancer: an in-silico & in-vitro investigation
Understanding the mechanism of action of anticancer agent plays a key role in effective clinical application of natural pr...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
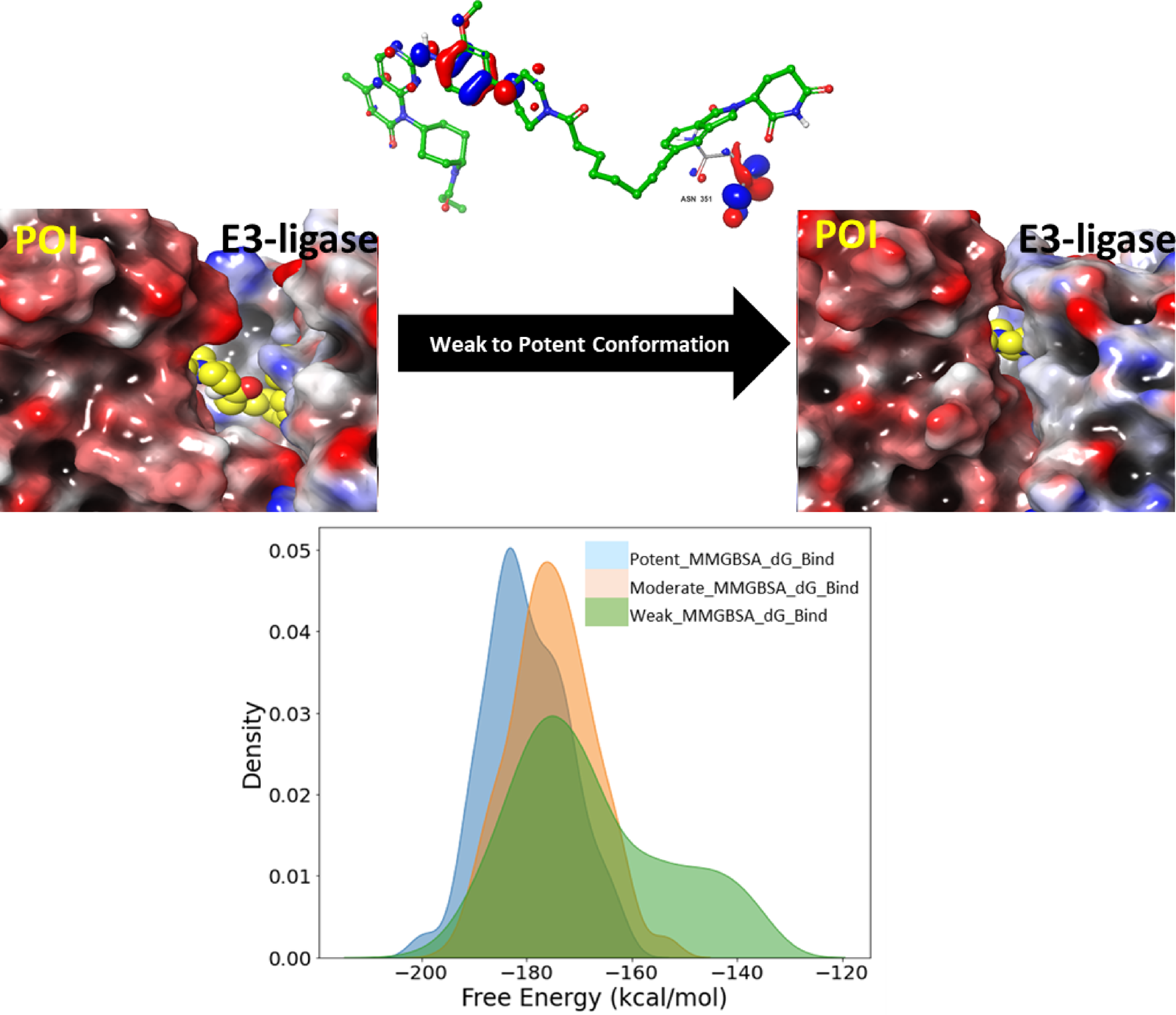
Mechanistic insights into PROTAC-mediated degradation through an integrated framework of molecular dynamics, free energy landscapes, and quantum mechanics: A case study on kinase degraders
Targeted protein degradation by proteolysis-targeting chimeras (PROTAC) is dependent on formation and plasticity of ternar...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
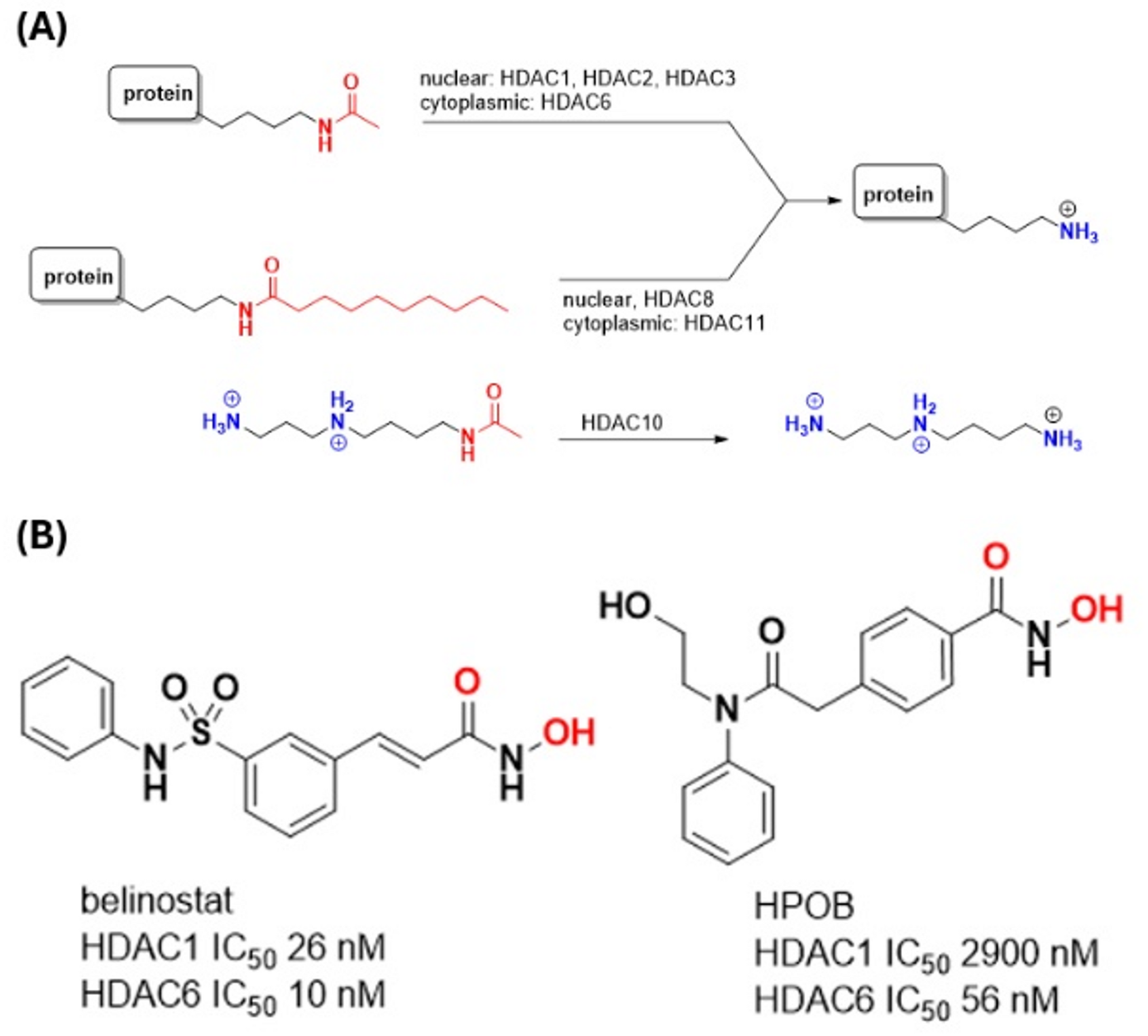
Protein domain movement involved in binding of belinostat and HPOB as inhibitors of histone deacetylase 6 (HDAC6): a hybrid automated-interactive docking study
DockIT is a tool for interactive molecular docking that can model both the local and global conformational response of the...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
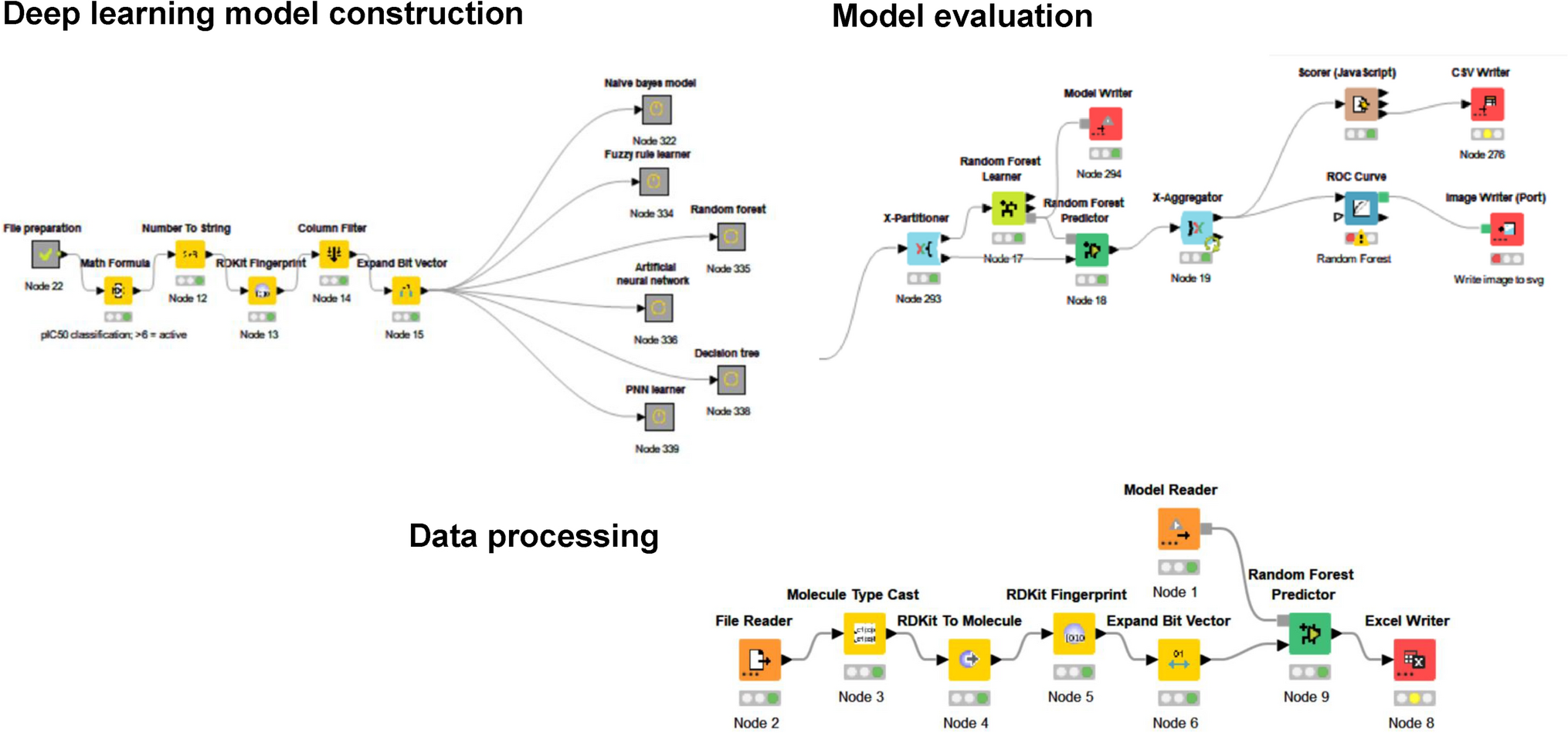
Integrated machine learning and deep learning-based virtual screening framework identifies novel natural GSK-3β inhibitors for Alzheimer’s disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder lacking effective therapies. Glycogen synthase ...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
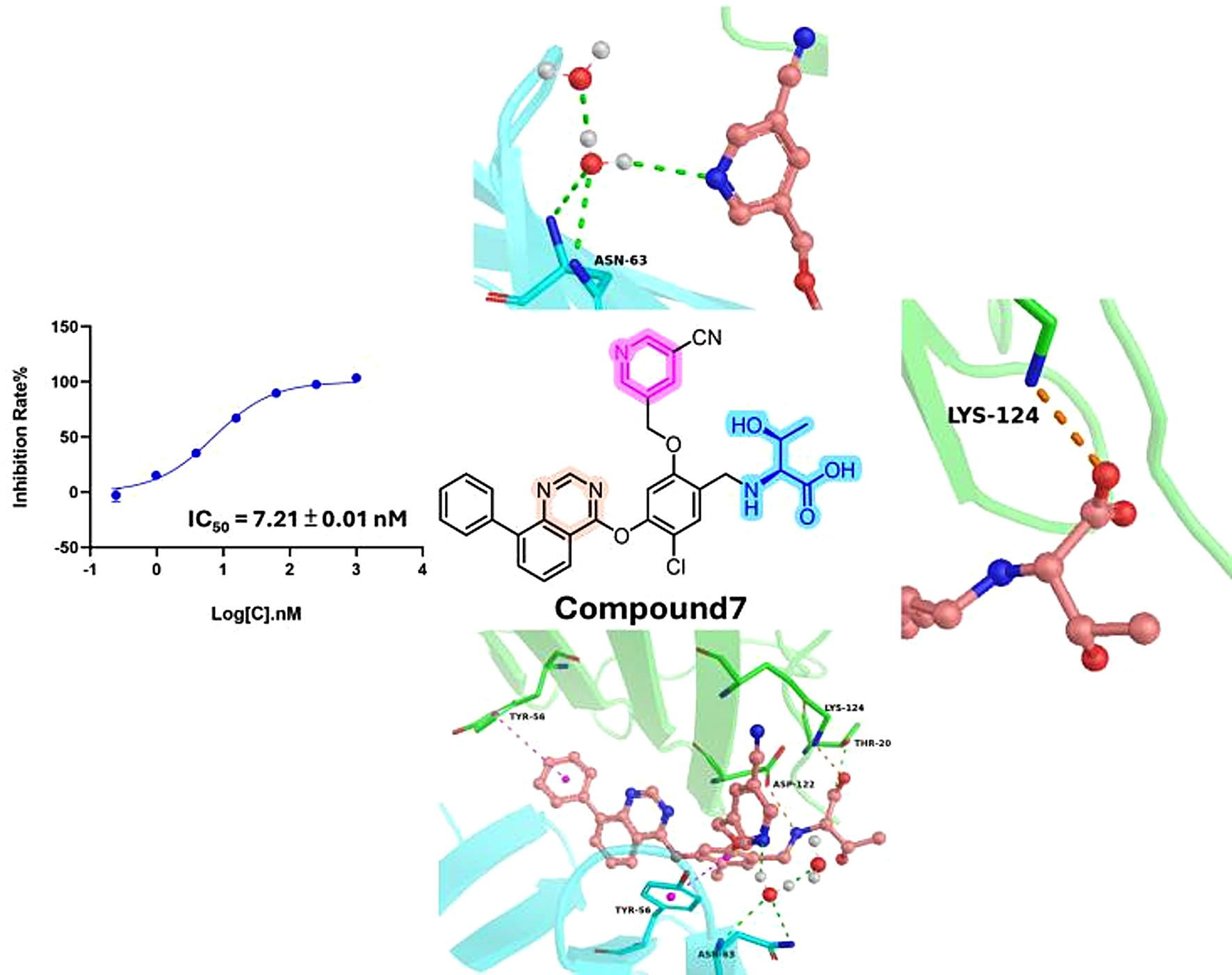
Design, synthesis, evaluation and molecular modeling of quinazoline derivatives bearing amino acids as small-molecule PD-L1 inhibitors
Herein, we reported a series of quinazoline derivatives bearing amino acids by introducing a rigid pyrimidine structure be...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
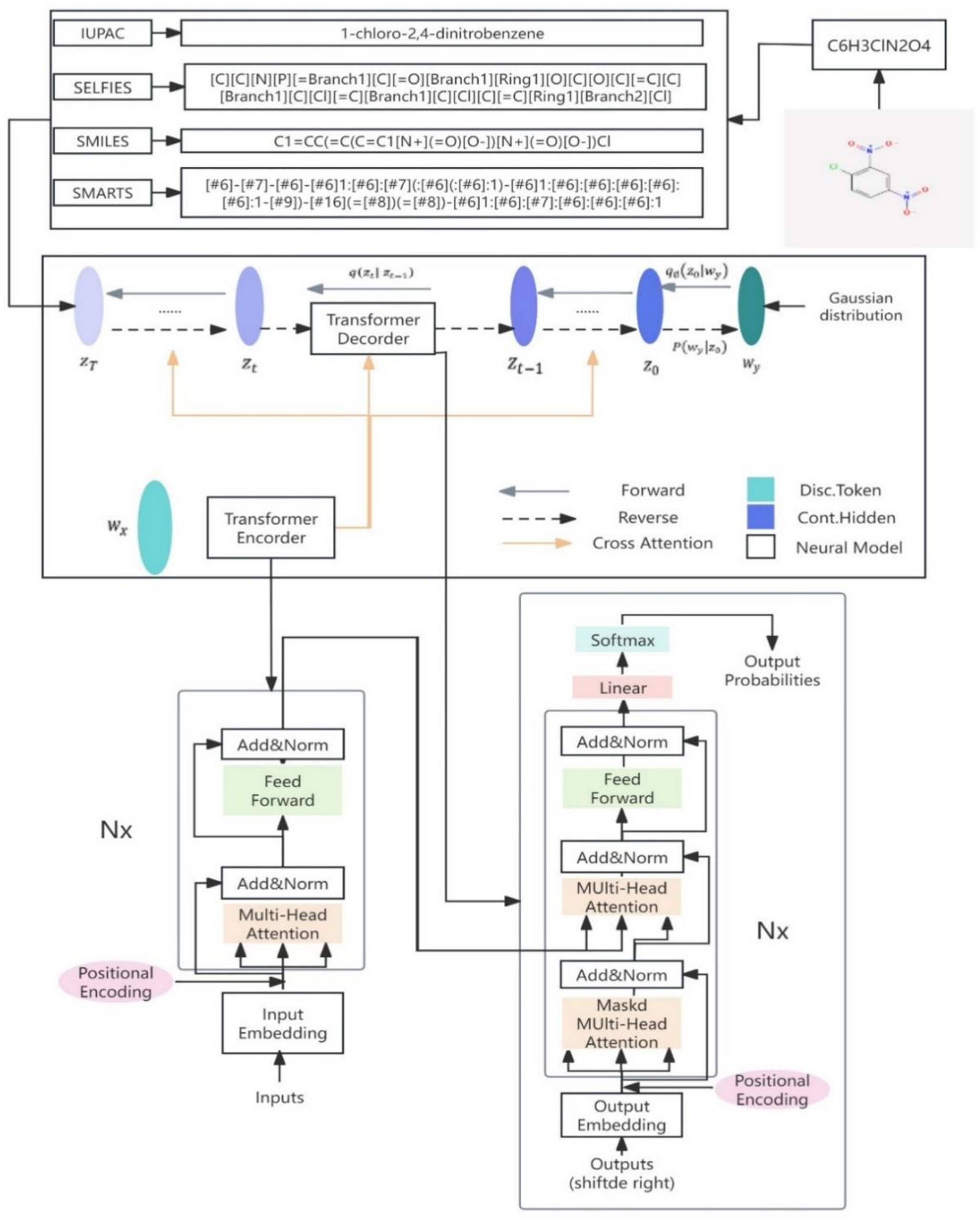
Comparison study of dominant molecular sequence representation based on diffusion model
In recent years, the emergence of large language models (LLMs), particularly the advent of ChatGPT, has positioned natural...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Anti-inflammatory potential of from : insights from molecular docking, ADMET, DFT, and studies
Grewia optiva, a medicinal plant native to northern Pakistan, has traditionally been valued for managing pain and inflamma...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Molecular dynamics simulations reveal mechanistic insights into aptamer-induced structural rearrangements in viral capsid proteins
Macrobrachium rosenbergii nodavirus is a major viral pathogen responsible for white tail disease in giant freshwater prawn...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
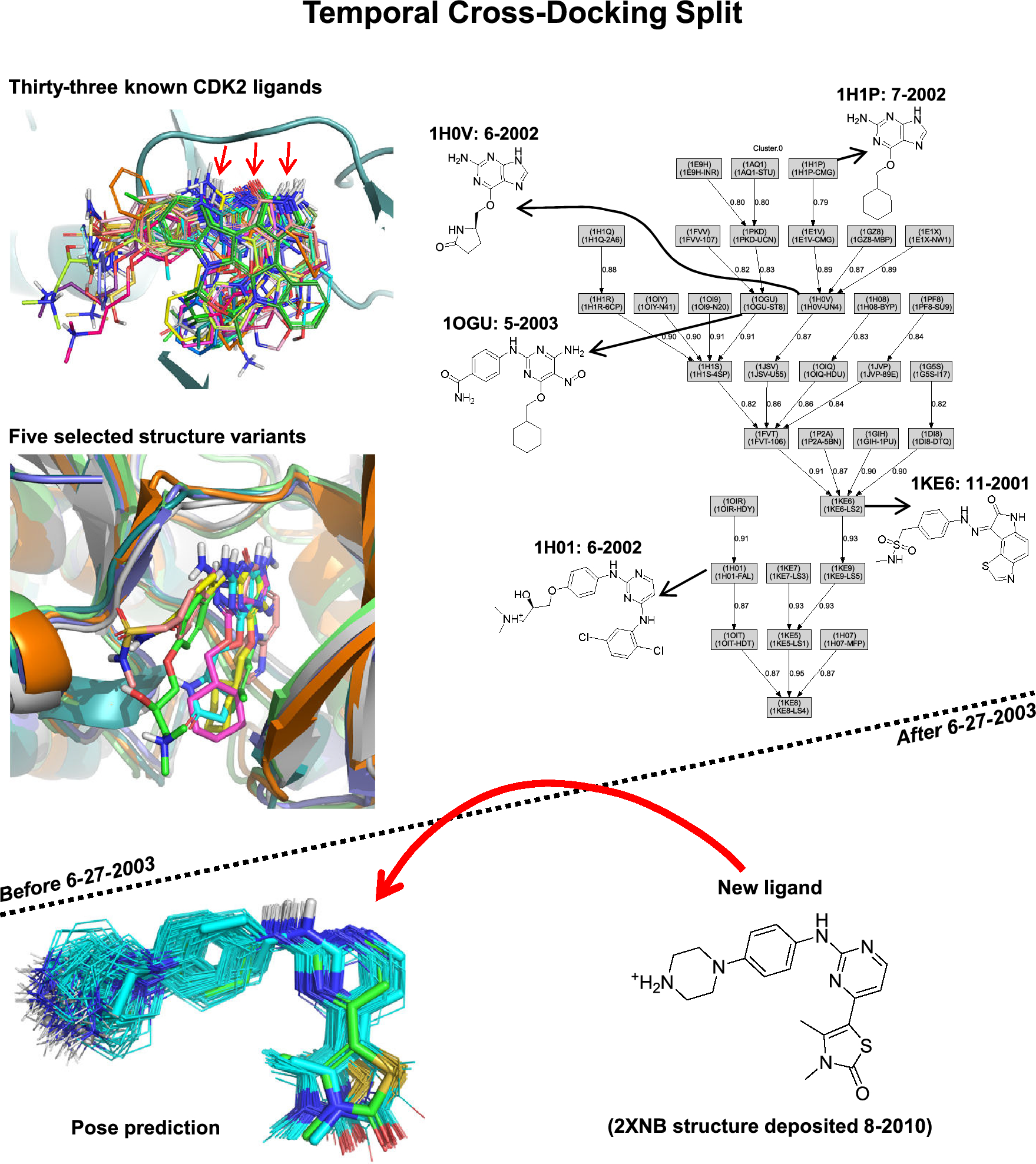
Structure-based pose prediction: Non-cognate docking extended to macrocyclic ligands
So-called “cross-docking” is the prediction of the bound configuration of small-molecule ligands that differ f...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Steered molecular dynamics simulation as a post-process to optimize the iBRAB-designed Fab model
Therapeutic monoclonal antibodies are an effective method of treating acute infectious diseases. However, knowing which of...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Comparative assessment of physics-based in silico methods to calculate relative solubilities
Relative solubilities, i.e. whether a given molecule is more soluble in one solvent compared to others, is a critical para...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
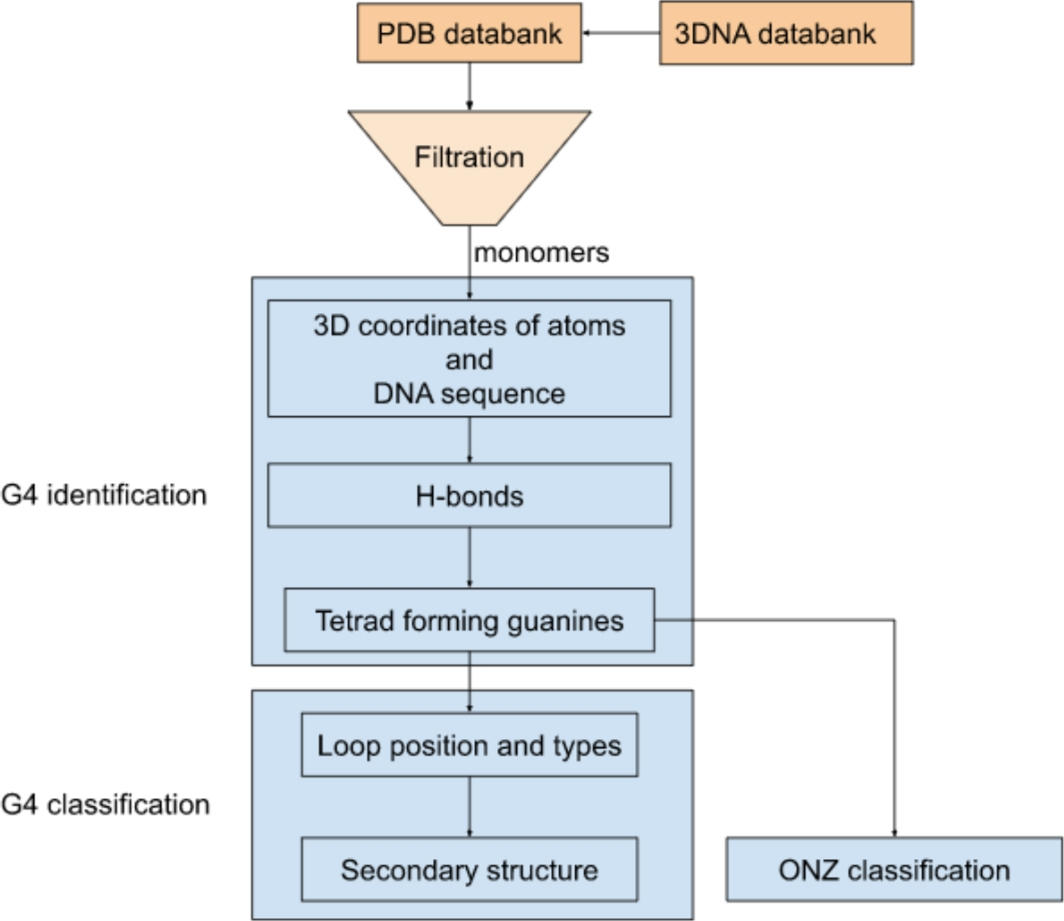
Computational Identification and Illustrative Standard for Representation of Unimolecular G-Quadruplex Secondary Structures (CIIS-GQ)
G-quadruplexes refer to a large group of nucleic acid–based structures. In recent years, they have been attracting a...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
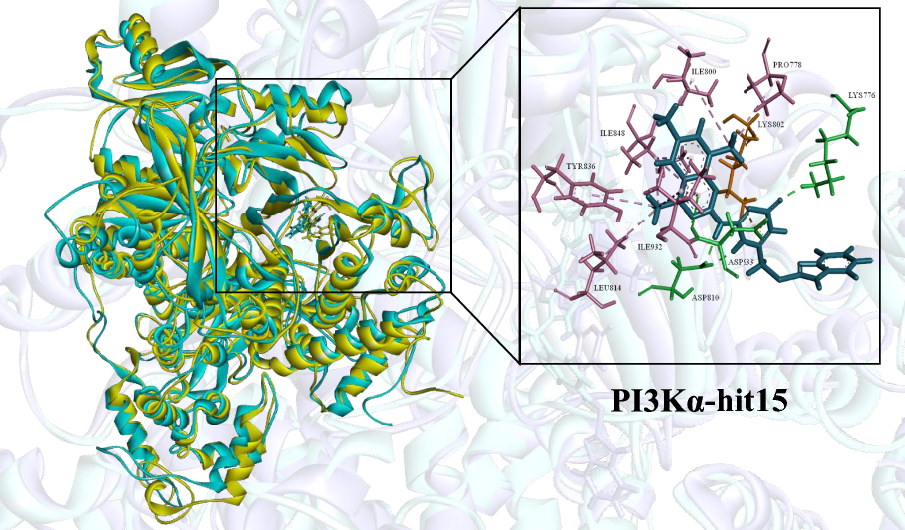
Identification of novel inhibitors targeting PI3Kα via ensemble-based virtual screening method, biological evaluation and molecular dynamics simulation
PIK3CA gene encoding PI3K p110α is one of the most frequently mutated and overexpressed in majority of human cancers....
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
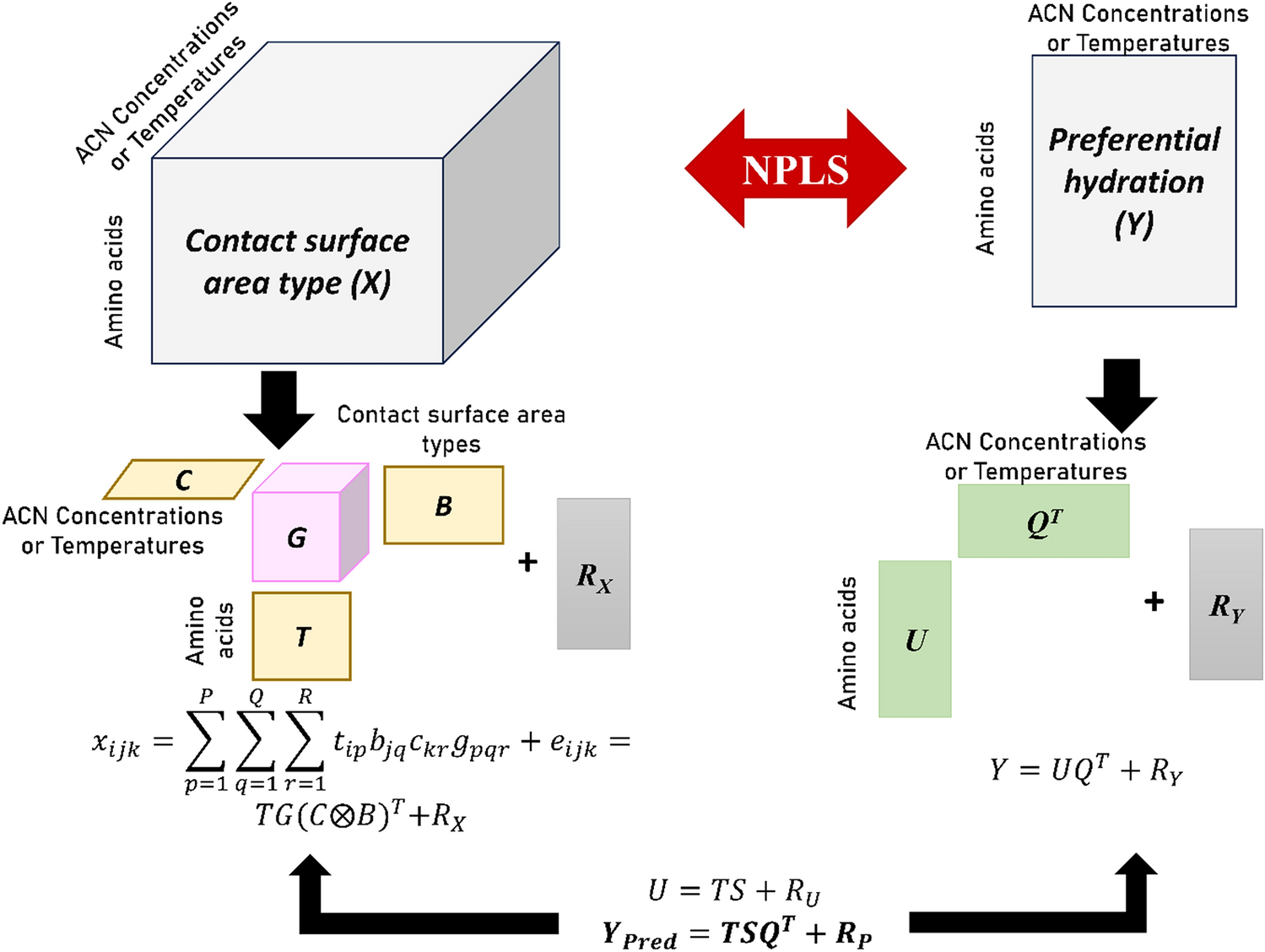
Understanding the relationship between preferential interactions of peptides in water-acetonitrile mixtures with protein-solvent contact surface area
The influence of polar, water-miscible organic solvents (POS) on protein structure, stability, and functional activity is ...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
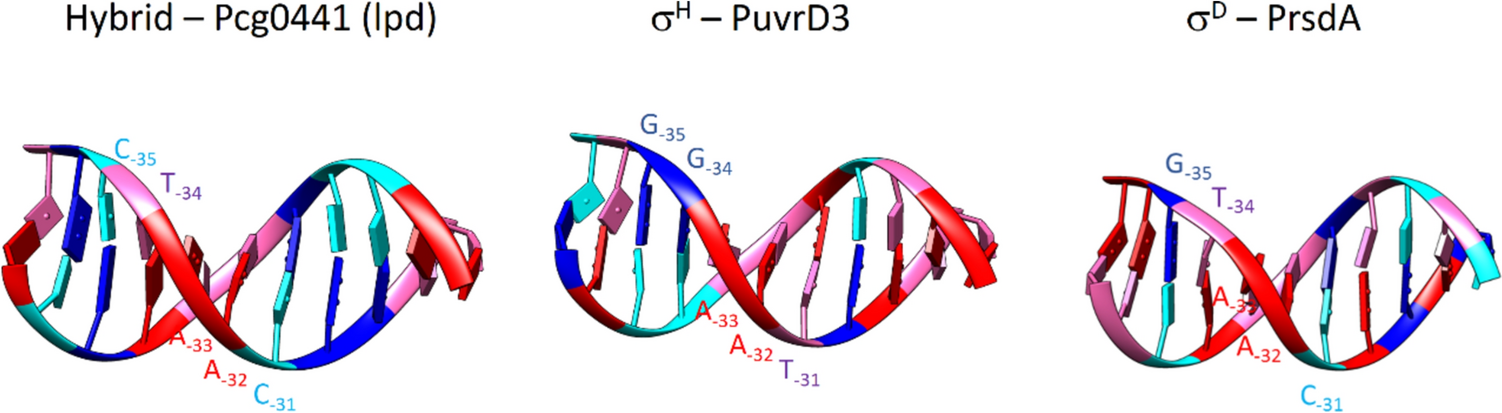
Promoter recognition specificity of Corynebacterium glutamicum stress response sigma factors σD and σH deciphered using computer modeling and point mutagenesis
This study aimed to reveal interactions of the stress response sigma subunits (factors) σD and σH of RNA polymer...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
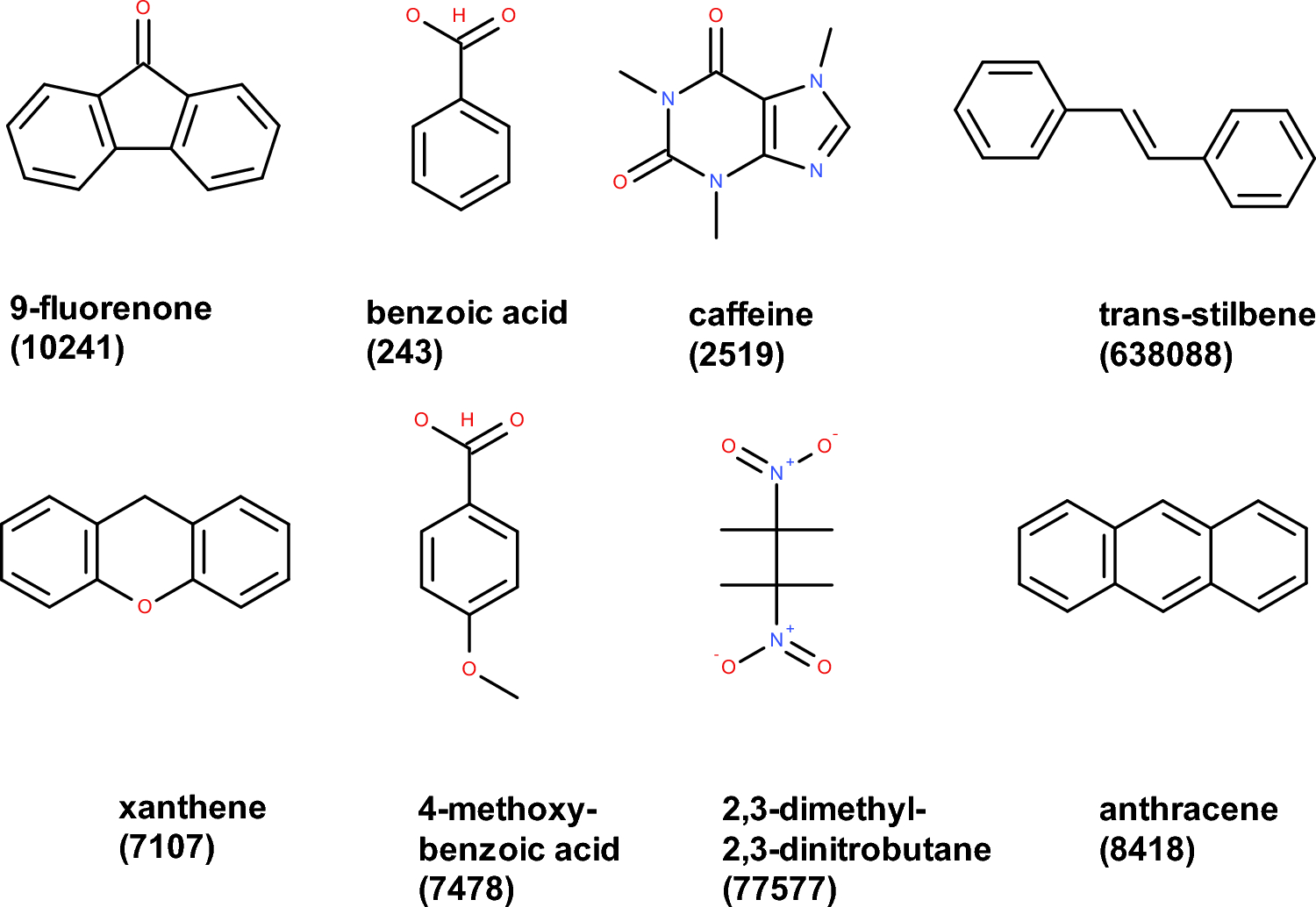
Combining crystallographic and binding affinity data towards a novel dataset of small molecule overlays
Although small molecule superposition is a standard technique in drug discovery, a rigorous performance assessment of the ...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
MolGraph: a Python package for the implementation of molecular graphs and graph neural networks with TensorFlow and Keras
Molecular machine learning (ML) has proven important for tackling various molecular problems, such as predicting molecular...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
User-centric design of a 3D search interface for protein-ligand complexes
In this work, we present the frontend of GeoMine and showcase its application, focusing on the new features of its latest ...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Load More
Modal title
×
Modal title
×
Share
Login
Global News and Health Forum
Join Now!
Member Login
Remember me
Forgot password?
Or using
Linkedin