
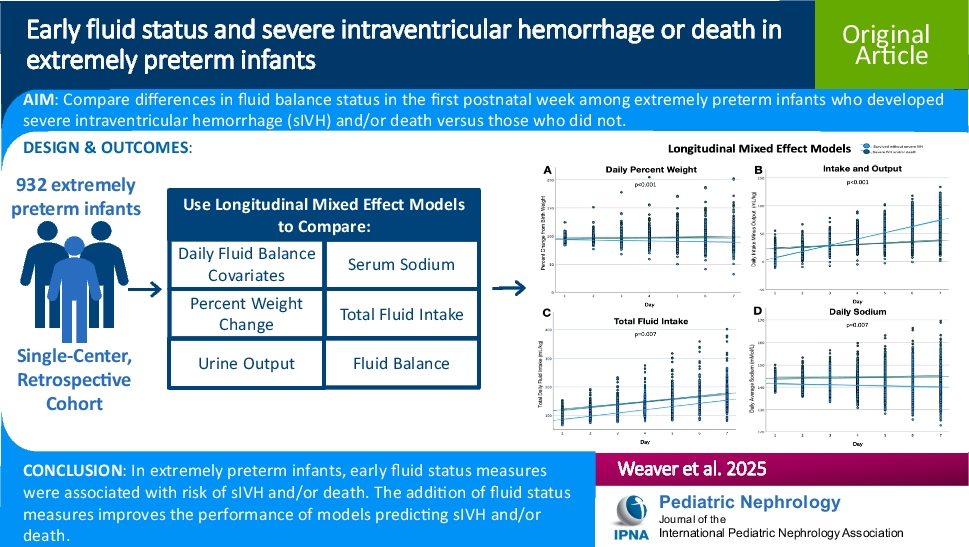
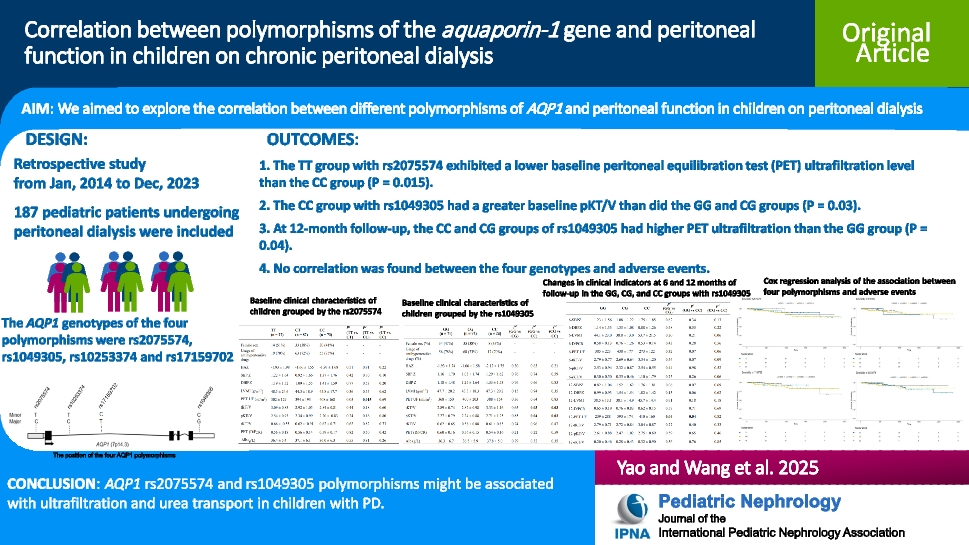
Remember me


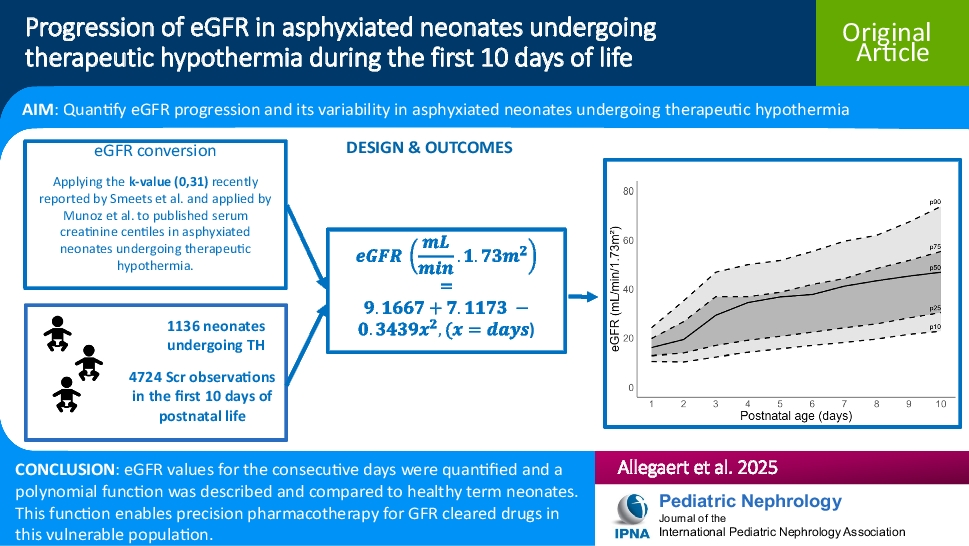
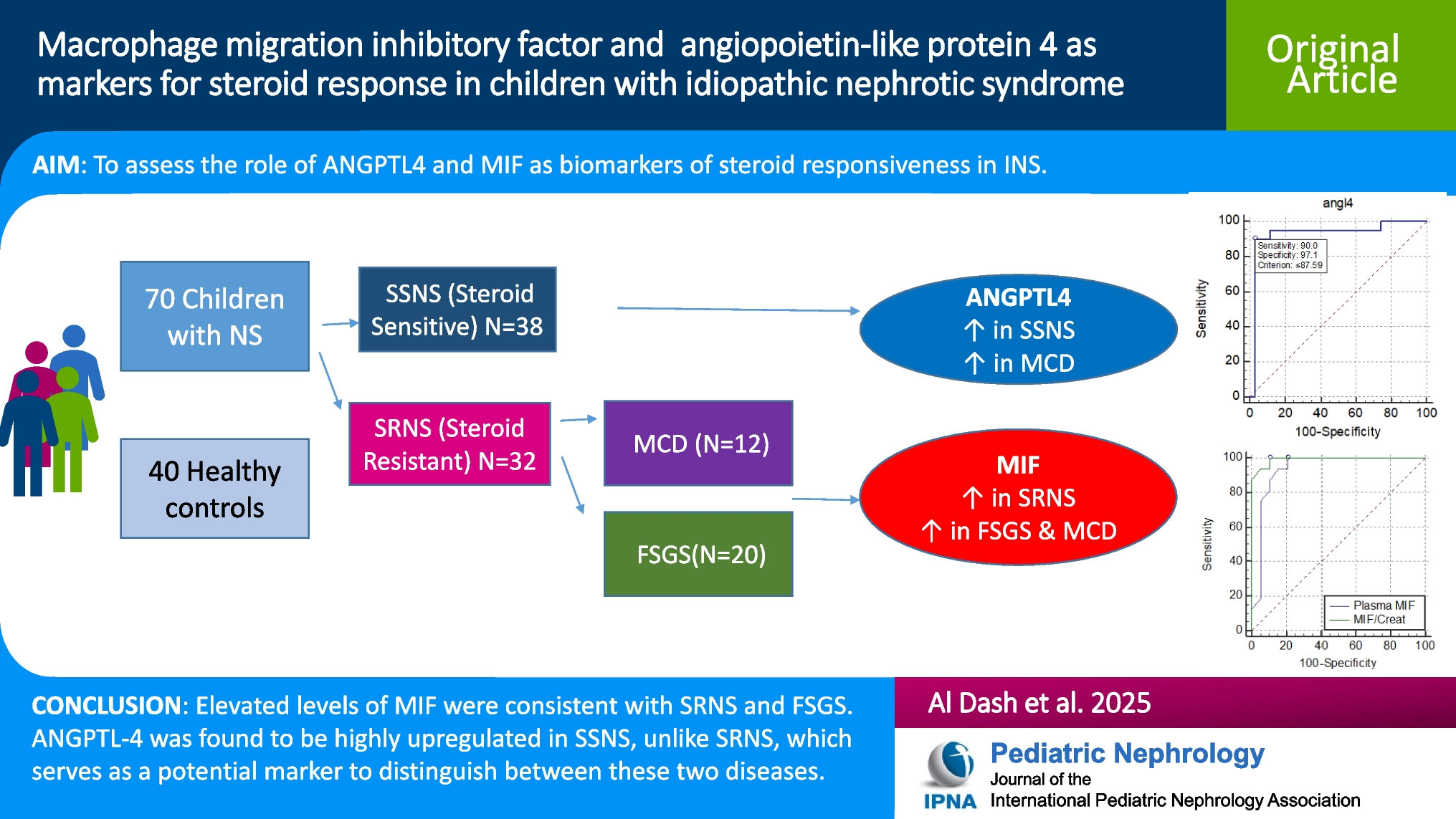
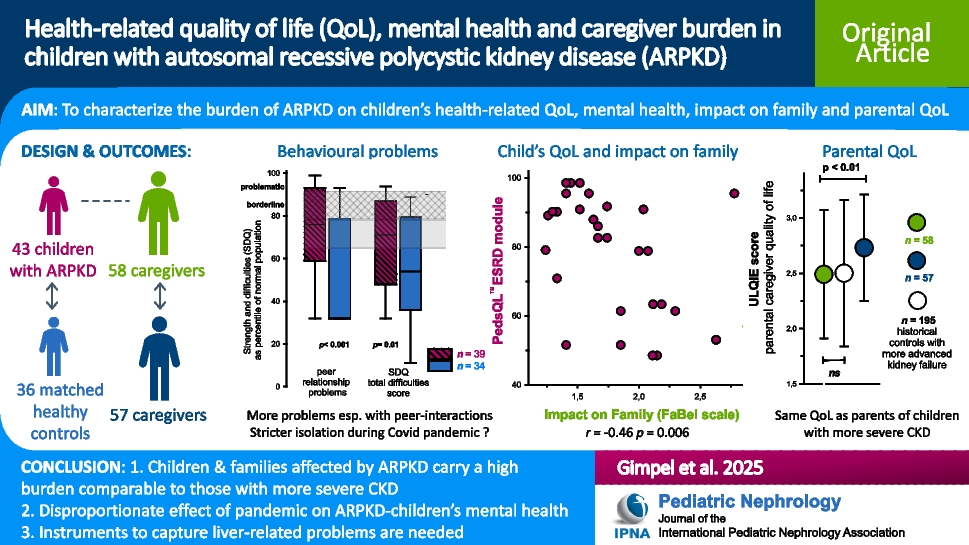

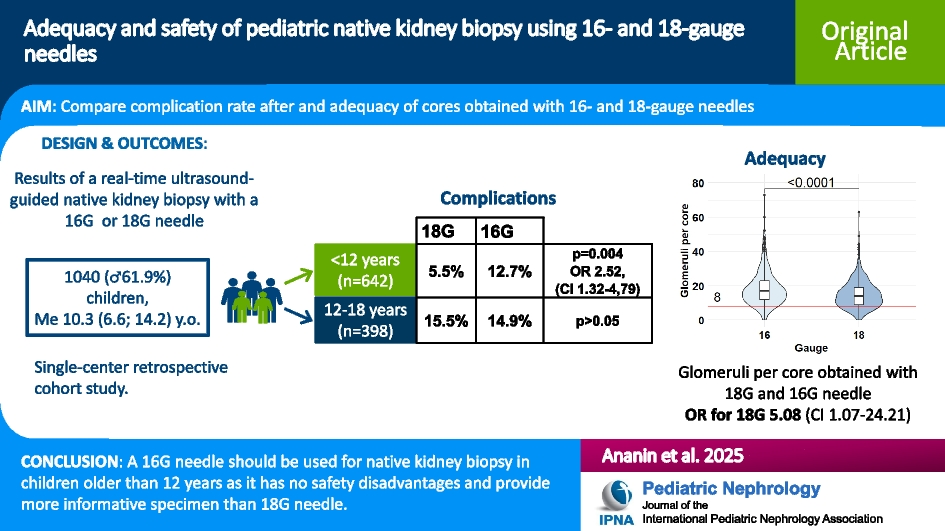

A 12-year-old Hispanic female with well-controlled systemic juvenile idiopathic arthritis (sJIA) complicated by hypertension, posterior reversible encephalopathy syndrome, and acute anterior uveitis, on biweekly tocilizumab, was referred to the nephrology clinic for persistent proteinuria. She was diagnosed with sJIA in 2022 and was in clinical remission on tocilizumab several months later. Proteinuria was first noted in 2023, 1.5 years after diagnosis of sJIA and 11 months into a state of clinically inactive disease.
She was referred to nephrology for proteinuria in 2024 with urine protein-to-creatinine ratio (UPC) ranging from 1 to 1.5 mg/mg (normal < 0.2 g/g) and a normal albumin of 4 g/dL. She had no microscopic hematuria, and her serum creatinine was normal at 0.49 mg/dL with an estimated glomerular filtration rate (eGFR) of 104.5 mL/min/1.73 m2 using the CKiD U25 equation. She was normotensive with a blood pressure of 99/70. Serologic workup was largely unremarkable aside from a positive antinuclear antibody titer (ANA) at 1:640 and a mildly low C3 level at 73 mg/dL (reference range 93–203 mg/dL). At the time of presentation with proteinuria, the patient’s sJIA was clinically inactive, with no fevers, rash, or arthritis, and normal inflammatory markers (ESR/CRP).
Initial kidney biopsy revealed mild C3GN versus latent infection-related GN. The sample contained 15–22 glomeruli, of which 1–3 were globally sclerosed. The remaining glomeruli showed mild segmental mesangial hypercellularity. Focal mild interstitial fibrosis and tubular atrophy (IFTA) were seen without significant interstitial inflammation. IF microscopy showed 5 glomeruli, 1 of which was globally sclerosed. Tubulointerstitial changes were similar to LM findings noted above. IF showed mesangial staining for C3 (1–2 +). No glomerular staining for IgG, IgA, IgM, C1q, or kappa or lambda light chains was seen. EM revealed parts of 3 glomeruli, 2 of which were globally sclerosed. A few mesangial electron-dense deposits and a single subepithelial electron-dense deposit were seen and identified as normal. The podocyte foot processes showed focal effacement. Serum C3 normalized on repeat testing one month later to 99.6 mg/dL. The C3 Glomerulopathy Complement Panel (C3G-CP) was sent to Iowa Health Care and was negative.
She was started on angiotensin-converting enzyme inhibitor (ACE-I) therapy for two months, yet UPC increased into the nephrotic range (2–2.2 mg/mg). High-dose prednisone (60 mg daily) was initiated for 4 weeks, resulting in mild improvement of her UPC to 1.4 mg/mg. Steroids were tapered, and mycophenolate mofetil (MMF) (600 mg/m2 twice daily) was added alongside daily ACE-I. Tocilizumab was spaced to every six weeks after MMF initiation.
Despite induction immunosuppression directed at C3GN, she had persistent proteinuria with UPC of 1.75 mg/mg; therefore, a second biopsy was performed. Results showed features of membranous glomerulopathy with masked IgG-kappa deposits. LM revealed thickened glomerular basement membranes with mild mesangial hypercellularity, raising suspicion for membranous nephropathy. Jones silver stain demonstrated frequent glomerular basement membrane (GBM) spikes, and several glomeruli showed segmental sclerosis. Again, no significant IFTA was noted. Routine IF again demonstrated only granular mesangial C3 staining. Given suspicion for MGMID based on her personal history of an autoimmune disease, IF was repeated on paraffin-embedded tissue with protease digestion, which revealed IgG-kappa deposits (Fig. 1).
Fig. 1
A Thickening of the glomerular basement membranes with abundant GBM “spikes” and “vacuoles” (Jones silver stain, × 400). B By routine immunofluorescence on fresh/frozen tissue, the glomeruli are negative for IgG and kappa and lambda light chains (× 400). C Following protease digestion of the formalin-fixed, paraffin-embedded tissue, direct immunofluorescence shows positive granular capillary wall staining for IgG and kappa light chain, and negative staining for lambda light chain (× 400). D Electron microscopy reveals numerous subepithelial immune-type electron-dense deposits with frequent GBM “spike” formation (× 4000)
Retrospective paraffin IF staining of the first biopsy was negative. EM from the initial biopsy showed only infrequent subepithelial deposits, likely suggesting early disease. At this time, we elected to initiate therapy with steroids and MMF for its efficacy and relative simplicity, as it does not require routine therapeutic drug monitoring. Additionally, we aimed to avoid the potential burden of an additional infusion on her quality of life.
This patient has remained on ACE-I and MMF with progressive improvement, with UPC decreasing to 0.32 mg/mg after 7 months (Fig. 2). Creatinine at last follow-up was 0.47 mg/dL. Although it is unknown what her response would have been without intervention, we hypothesize that the continued resolution of her proteinuria is a result of time on treatment. Her kidney function remains normal, and tocilizumab infusions have been discontinued as her sJIA has been in long-term remission.
Fig. 2
Trend in proteinuria over time in a patient with membranous-like glomerulopathy with masked IgG-kappa deposits (MGMID). Proteinuria was measured at multiple time points, beginning at initial presentation and continuing through kidney biopsy and initiation of various treatments, including an ACE inhibitor, corticosteroids, and mycophenolate mofetil
Comments (0)