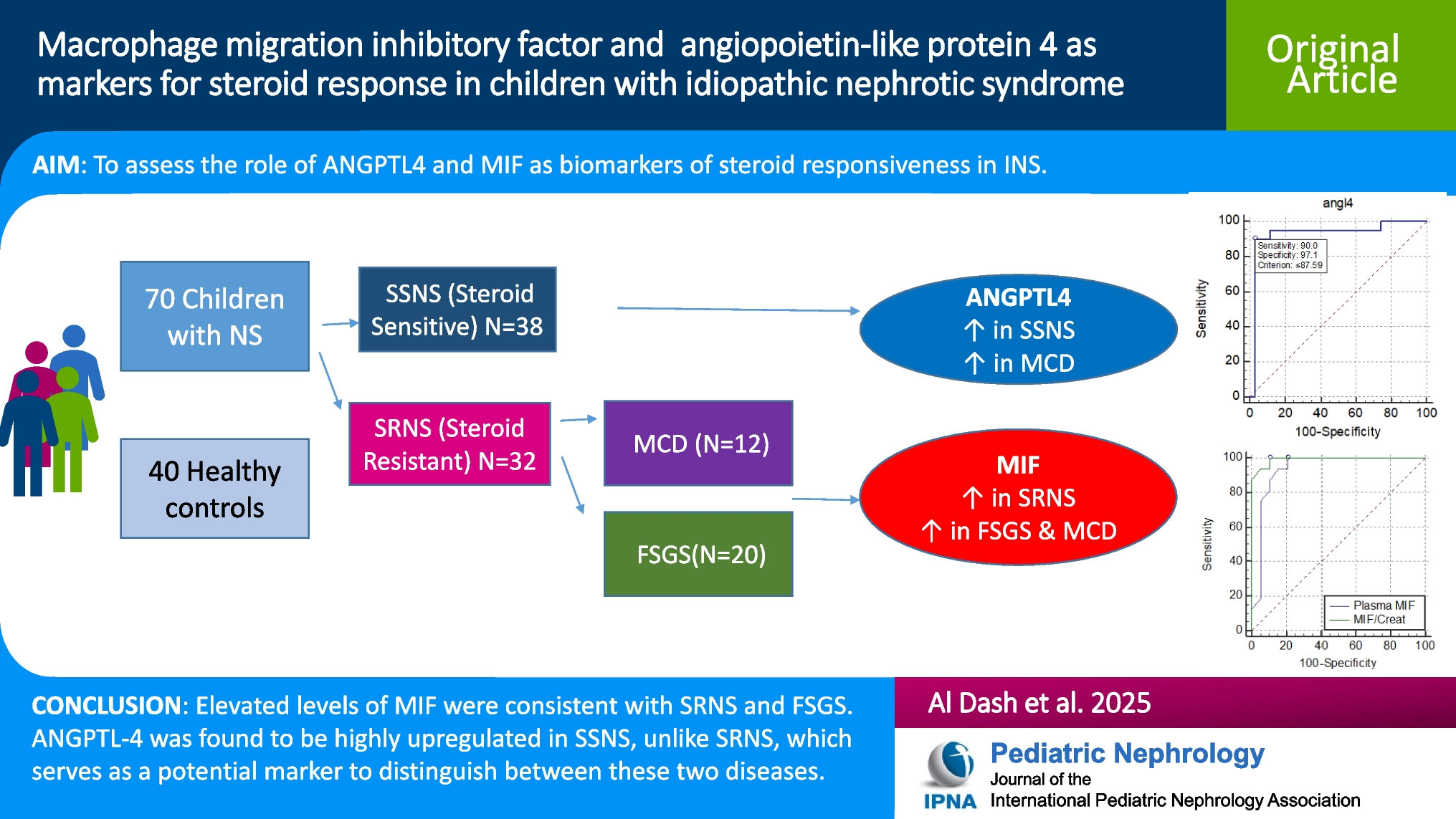
Idiopathic nephrotic syndrome is the predominant kind of NS, with the remaining etiologies being secondary to other conditions such as congenital, infections, or glomerular diseases. There are numerous theories in the literature on the pathogenesis of INS, attributed to various mechanisms such as glomerular abnormalities, cytokines, or immunological problems [12]. In this case-control study, we wanted to study the role of serum MIF and urinary MIF as predictive markers for INS steroid responsiveness, as well as their utility in understanding the underlying pathology, steroid resistance mechanism, and potential therapeutic targets. In terms of serum MIF, we found a significantly elevated level in the SRNS group relative to the SSNS group (p = 0.001). Cuzzoni et al. reported that serum MIF levels were markedly elevated in the setting of steroid-resistant INS, with a mean concentration of 759.7 pg/mL, compared to steroid-sensitive INS and steroid-dependent INS, with a concentration of 414.1 pg/mL (p = 0.022). Concurring with our findings, they proposed that serum MIF could be of use as a biomarker to predict responsiveness to steroid therapy in children with INS [5]. Ramayani et al. also put forth data supporting our proposition, as they found that children who had SRNS had a genetic mutation in the gene MIF-173G/C, with the C allele being frequently met in the SRNS group relative to the SSNS and healthy controls (p = 0.025), and they also noted that serum MIF levels were significantly increased above 20 ng/mL in the SR group (p = 0.04) as well as in the group with the C allele (p = 0.04), which suggests that the variability in expression of MIF may have a genetic basis and may determine response to steroid in INS [13]. In contrast to our findings, Zwiech et al. concluded that levels of serum MIF were, indeed, significantly increased in subjects with glomerulonephritis (whether responsive or not to immunosuppressive treatment) in contrast to healthy controls (p = 0.036); however, the levels did not differ significantly between steroid-responsive and steroid-resistant cases, whether at baseline or after initiation of immunosuppressive treatment [14]. Our findings can be attributed to the fact that MIF is a proinflammatory cytokine that induces the chemotaxis of macrophages and T cells, evoking an inflammatory response, adding to the fact that it antagonizes the immunosuppressive effect of glucocorticoids—despite being released by glucocorticoids during a stressful kidney event, such as acute kidney injury—thus, it causes an inflammatory process in the kidney leading to activation of macrophages, which subsequently causes the release of more MIF, and a vicious cycle is established [15,16,17]. Therefore, patients who are impacted by steroid-resistant kidney disease may have favorable outcomes if a blocking agent targeting MIF is used instead of glucocorticoids [18]. Reflecting on urinary MIF, we demonstrated a significant rise in the SRNS group as opposed to the SSNS group (p = 0.035). This could be explained by the fact that the diseased kidney tissues express MIF at dramatic levels, which raises the serum levels of MIF, and consequently, due to the underlying glomerulonephritis, a urinary output impairment ensues, leading to higher excretion of MIF in the urine. Zwiech et al. supported this finding as they observed that urinary MIF was substantially higher in primary glomerulonephritis versus healthy controls, irrespective of the type of primary GN (p = 0.006, p = 0.009). They also noted that following treatment, the levels of urinary MIF were significantly greater in the steroid non-responders than in those who responded to treatment (p < 0.05) [9]. Endorsing our findings, Lan et al. discerned that, according to immunohistochemical analysis, MIF was a normal constituent of kidney tissues but highly upregulated in the case of glomerulonephritis, and they inferred that kidney expression of MIF was much more prominent in areas of tissue damage when compared with the histochemical appearance of other areas without damage, and they owed that to macrophage (p < 0.0001) and T cell (p < 0.05) release of MIF at areas of focal damage, not to the normally expressed MIF by glomerular or tubular cells of the kidney. Overall, they substantiated our data by confirming that elevated urinary MIF was, in fact, mainly due to active inflammation and that urinary MIF could serve as a potential marker of kidney inflammation and a potential therapeutic target in patients who fail to respond to steroids [19]. Others, such as Kong et al., reported ambivalent results that contrast with our results; Kong et al.’s results hint that the elevated urinary MIF levels may be attributed to MIF expression by intrinsic cells of the kidney (podocytes, tubular cells, endothelial cells, and mesangial cells), along with resident macrophages, T lymphocytes, and fibroblasts [20]. Additionally, the SRNS group had a significantly greater MIF/creatinine ratio (p < 0.001).
The ROC analysis of our data regarding serum MIF showed that at a cutoff score of > 490 pg/ml, serum MIF could discriminate between SSNS and SRNS with a sensitivity of 100% and a specificity of 87% (AUC = 0.936). Our analysis and deductions are supported by Cuzzoni et al., who observed that patients with SRNS had higher levels of serum MIF and concurred that at a cutoff of > 501 pg/ml, MIF could significantly identify resistance to steroid therapy with a sensitivity of 85.7% and a specificity of 71.4% [5]. In our study, we intended to evaluate the role of ANGPTL-4, a protein expressed in various tissues of the body, including endothelial cells and podocytes, in the context of INS. Intriguingly, we noted that serum ANGPTL-4 levels were significantly higher in the SSNS group (186.67 pg/ml) as opposed to the SRNS group (73.46 pg/ml, p = 0.000), but insignificantly higher than in the control group. Li et al. experimented on nephrotic mice, comparing those without the ANGPTL-4 gene (using the CRISPR/Cas9 technique to eliminate the ANGPTL-4 gene) with mice that still had the gene, in terms of proteinuria and hyperlipidemia. They found that lipid disorders and podocyte effacement were relieved in mice with the knocked-out ANGPTL-4 gene, with significantly lower proteinuria and substantially reduced total cholesterol and triglyceride levels as opposed to nephrotic mice with the gene, underlining the role of ANGPTL-4 in both the pathophysiology of NS and the associated hyperlipidemia. Furthermore, they knocked out the gene in the ANGPTL4 +/+ mice, which then displayed notable relief of severe proteinuria and amelioration of disordered lipid metabolism [21]. Following our results, Clement et al. addressed the function of ANGPTL-4 in nephrotic syndrome by injecting ANGPTL4 +/+ rats with a nephrotoxic agent, and they noted a significant elevation in the levels of ANGPTL4 mRNA in these rats. However, when ANGPTL4-/- rats were injected with the same nephrotoxic agent, the levels of proteinuria and podocyte effacement were substantially lower. Moreover, they studied the possible contribution of sialylation in the variable response of tissues to ANGPTL-4 and showed that nephrotic rats that were treated with ManNAc (a sialylating agent) showed much lower levels of proteinuria compared to baseline. In addition to that, they interrogated the sensitivity of ANGPTL-4 to glucocorticoids and concluded that this characteristic significantly favors MCD and other SSNS, as glucocorticoids seem to significantly increase the expression of the sialylated form of ANGPTL-4, leading to amelioration of proteinuria [9]. This could be explained by the fact that ANGPTL-4 acts on various tissues of the body in multiple ways, some of which induce podocyte injury and proteinuria and inactivation of lipoprotein-lipase (LPS) [22]. A hyposialylated form of ANGPTL-4 is expressed exclusively by podocytes of the glomeruli, inducing proteinuria. This hyposialylated form then moves into the bloodstream, which is then expressed, after being sialylated, by other tissues like adipose tissue, cardiomyocytes, and skeletal muscles, and with treatment with glucocorticoids in steroid-sensitive nephrotic syndrome, this form of ANGPTL-4 is expressed even more by peripheral tissues [23]. The sialylated form of ANGPTL-4 then binds to glomerular endothelial cells and reduces proteinuria but also inhibits LPS, which causes hyperlipidemia [24]. This explains the partial response to steroids in some types of nephrotic syndrome, as well as the steroid-dependent types. This approach in treatment leads to activation of the systemic and local feedback loop of ANGPTL-4 expression, which then worsens hyperlipidemia of nephrotic syndrome and leads to unsatisfactory responsiveness of proteinuria [25]. We further evaluated the treatment groups again after subgrouping based on the type of nephrotic syndrome into the FSGS group and MCD group. We determined that there were no statistically significant differences between both groups regarding all parameters except ANGPTL-4 levels, which were higher in the MCD group (184.55 pg/ml) in contrast to 73.46 pg/ml in the FSGS group. We deemed this to be of statistical significance (p < 0.001). This aligns with the results of Clement et al., who also noted that podocytes in both human and animal model MCD expressed ANGPTL-4 much more than podocytes in other kidney diseases such as membranous nephropathy, with unchanged ANGPTL-4 levels in focal segmental glomerulosclerosis (FSGS). Furthermore, they noted that ANGPTL-4 oligomers could be found in the urine of patients with MCD, as well as upregulated in their serum, unlike those with FSGS. They also stated that ANGPTL-4, being a steroid-sensitive molecule, has a significant contribution to the development of MCD, as its expression by podocytes results in a filtration defect, which in turn leads to nephrotic-range proteinuria. To confirm these results, they experimented on NHPS2-ANGPTL4 transgenic rats and extrapolated that the ANGPTL-4, via complex interplay with the podocyte-GBM interface, results in foot process effacement, and that mutation of the NHPS2 gene, which encodes the podocyte protein podocin, results in a steroid-resistant variant of nephrotic syndrome, namely FSGS [9]. Li et al. also reported on the role of ANGPTL-4 in the development of INS and indicated that glomerular expression of ANGPTL-4 was only notable in MCD, unlike MSPGN and FSGS [26]. This can be explained by the fact that ANGPTL-4 is a glucocorticoid-sensitive protein that is heavily upregulated in the kidney as well as peripheral tissues in the context of steroid-sensitive kidney disease, and due to being enacted through interaction with podocyte integral proteins such as podocin, which are regulated by genes such as NHPS2, mutations of these genes might be responsible for steroid-resistant nephrotic syndrome like FSGS, which might explain the significantly lower ANGPTL-4 expression in FSGS compared with its upregulation in MCD [9, 27,28,29].
Study limitations
This study has several limitations, including the relatively small sample size and the inability to study more biomarkers due to the lack of funding. Also, we were unable to measure the markers at the onset of the first attack in all of the patients.





Comments (0)