
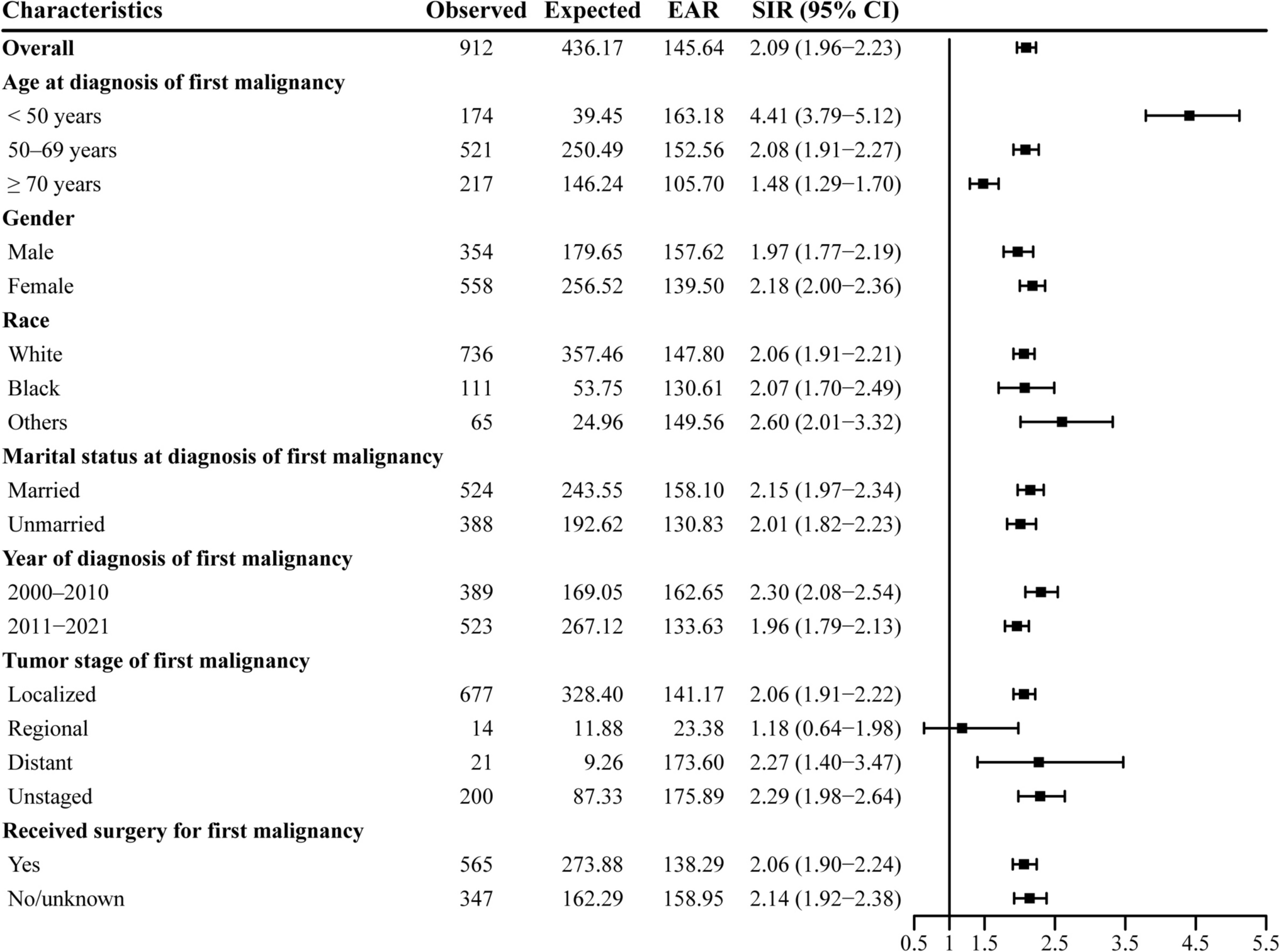
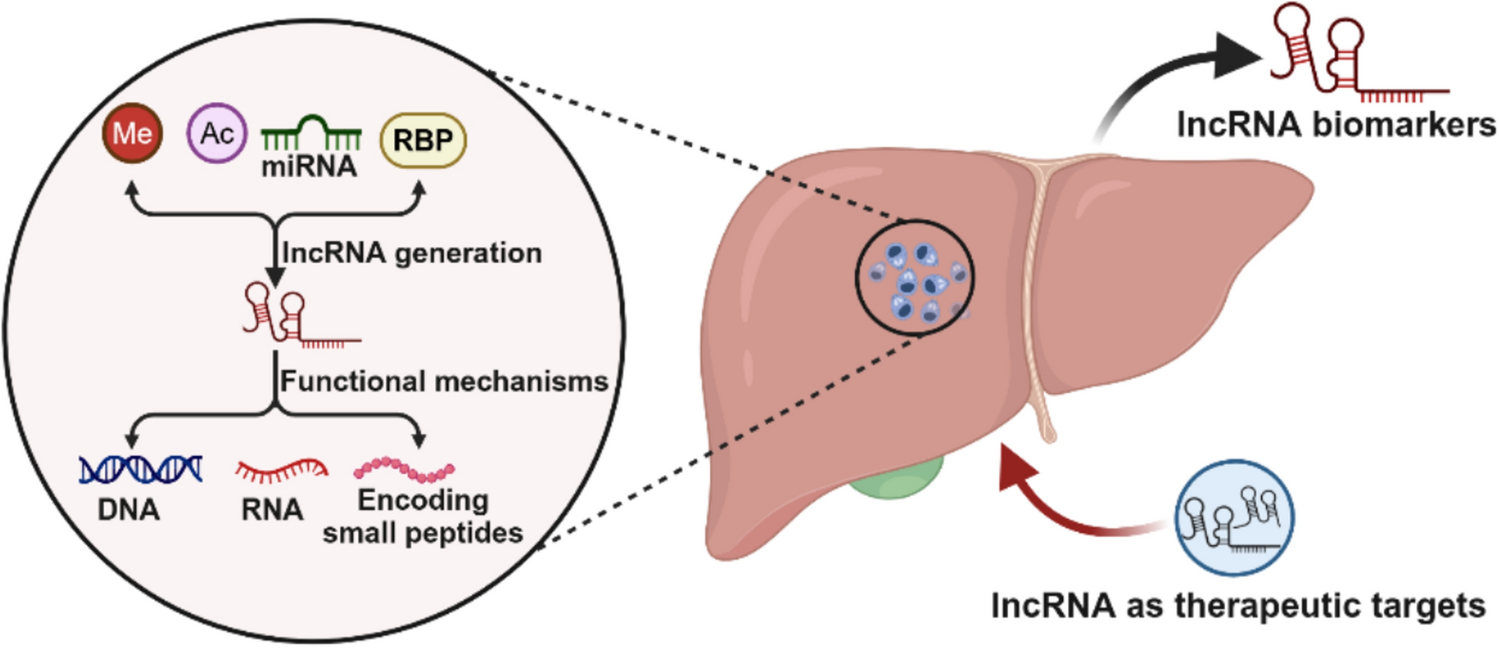
Remember me

This study aimed to explore the variation patterns of genes related to kinases in patients with HCC using the TCGA-LIHC cohort (Fig. 1). Out of 538 kinase-related genes, we found that 45 were expressed differentially between HCC and normal tissues. Specifically, the expression levels of 42 genes were upregulated in cancerous tissues, while 3 genes were downregulated, with all DEGs listed in Supplementary Table 3. Figure 2A shows a heatmap of the key kinase-related genes, while Fig. 2B displays a volcano plot. The results of the enrichment analysis revealed that the identified key genes exhibited significant enrichment in diverse biological functions associated with protein phosphorylation. These functions encompassed a spectrum of processes, notably protein phosphorylation, peptidyl-serine phosphorylation, protein autophosphorylation, and peptidyl-tyrosine phosphorylation. Furthermore, the enriched biological processes extended to cancer-related pathways, including the mitotic cell cycle, cell population proliferation, positive regulation of the cell cycle, PI3K-Akt signaling pathway, regulation of TP53 activity, and axon guidance (Fig. 2C). The protein–protein interaction network and MCODE components were identified in the key gene lists, as depicted in Fig. 2D. Furthermore, our study involved a thorough examination of genetic alterations in pivotal kinase-related genes among HCC patients from the TCGA cohort. This analysis resulted in the identification of the most prevalent mutations within the top 15 kinase-related genes. Remarkably, the PRKDC gene emerged as having the highest mutation frequency, recorded at 29.7%. In comparison, the mutation frequencies of the other genes in this group were found to range between 2.7 and 6.8%, as illustrated in Fig. 2E.
Fig. 1
A flow diagram of the research design
Fig. 2
The expression and variation of kinase-related genes in HCC patients. (A) Heatmap of the differentially expressed (DE) kinase-related genes between tumor and normal tissues. (B) Volcano plot of the DE-kinase-related genes in tumor and normal tissues (green: down-regulated DEGs; red: up-regulated DEGs; grey: unchanged genes). (C) Enrichment analyses based on the DE-kinase-related genes. (D) PPI network based on the DE-kinase-related genes. (E) An oncoplot of DE-kinase-related genes in the TCGA-LIHC cohort
Identification of molecular subtypes based on prognostic key kinase-related genesIn this study, univariate Cox regression analysis was employed to pinpoint kinase-related genes with prognostic importance for HCC patients within the TCGA dataset. This analysis successfully identified 27 genes significantly correlated with survival (P < 0.05), as listed in Supplementary Table 4. Using these genes, we classified patients from the TCGA-LIHC cohort into two distinct clusters. These clusters demonstrated robust internal consistency and stability, as evidenced in Fig. 3A–C. PCA further highlighted a marked differentiation between the clusters (Fig. 3D). Notably, the expression levels of the majority of these 27 key prognostic kinase-related genes were elevated in cluster 2 relative to cluster 1, as shown in Fig. 3E. Survival analysis indicated that patients in cluster 2 had a significantly poorer prognosis compared to those in cluster 1 (Fig. 3F).
Fig. 3
Clinical and immune characteristics of kinase-related subtypes. (A) Consensus clustering matrix when k = 2. (B) Consensus clustering CDF with k valued 2–9. (C) Relative change in area under the CDF curve for k = 2 through 9. (D) PCA analysis showed that HCC could be well differentiated into two subtypes based on the expression of prognostic key kinase-related genes. (E) Heatmap of the 27 prognostic key kinase-related genes between cluster 1 and cluster 2. (F) Kaplan–Meier curves of OS for two subtypes in HCC. The expression of immune cell infiltration (G) and immune/stroma score (H) between different subtypes. (I) The expression levels of immune checkpoint genes between different subtypes. *p < 0.05, **p < 0.01, ***p < 0.001
Immune characterization between different kinase-related subtypesCluster 1 and Cluster 2 displayed notable differences in various immune cell populations (Fig. 3G), encompassing B cells naive, B cells memory, T cells CD4 memory resting, T cells CD4 memory activated, T cells follicular helper, NK cells resting, macrophages M0/M2, and dendritic cells resting. Notably, Cluster 2 exhibited higher immune scores and lower stromal scores compared to Cluster 1 (Fig. 3H). Furthermore, an examination of the expression levels of six immune checkpoint genes, namely PDCD1, CD274, CTLA4, TIGIT, LAG3, and HAVCR2, revealed elevated expression in Cluster 2 relative to Cluster 1 (Fig. 3I).
Biological characteristics of kinase-related subtypesTo investigate the potential reasons for the different prognosis and immune characteristics observed between the two clusters, we examined the hallmark pathways enriched in each cluster. Using GSEA analysis, we found that cluster 1 was enriched in BILE_ACID_METABOLISM, XENOBIOTIC_METABOLISM, and FATTY_ACID_METABOLISM pathways (Fig. 4A). Additionally, cluster 1 exhibited upregulated expression of genes involved in DNA replication and cell cycle-related pathways, including E2F targets, G2M checkpoint, MYC targets, MTORC1, and PI3K-Akt pathway (Fig. 4A). In contrast, cluster 2 showed enrichment in cancer-related pathways such as PI3K_AKT_MTOR_SIGNALING and WNT_BETA_CATENIN_SIGNALING, as well as cell cycle-associated G2M_CHECKPOINT and E2F_TARGETS pathways (Fig. 4B). Furthermore, we identified 157 genes with differential expression levels between the two clusters (Supplementary Table 5), which were enriched in many biological functions and pathways, including cell cycle, Chemical carcinogenesis, p53 signaling pathway, Drug metabolism, and kinase activity (Fig. 4C, D).
Fig. 4
Biological characterization of kinase-related subtypes. The GSEA pathway enrichment analysis in cluster 1 (A) and cluster 2 (B). (C) GO enrichment analyses based on the prognostic DEGs. adjusted p < 0.05. BP, biological process; CC, cellular component; MF, molecular function. (D) KEGG enrichment analyses based on the prognostic DEGs. p < 0.05
Establishment and verification of a kinase-related gene signature in HCCEstablishment of kinase‐related gene signatureIn the initial phase of our analysis, we discerned 120 DEGs out of a total of 157, demonstrating a significant correlation with OS in HCC, as detailed in Supplementary Table 6. Subsequently, we employed LASSO Cox regression analysis to derive an 8-gene signature comprising UAP1L1, CENPA, TRIP13, PLGLA, CDCA8, PKIB, KIF20A, and SLC16A3 from these prognostic-kinase-related DEGs. Among the eight signature genes, seven were significantly upregulated in tumor tissues compared to normal tissues in the TCGA liver cancer dataset, while PLGAL was downregulated (Fig. S1). To validate these findings, we collected 20 pairs of clinical HCC and adjacent normal tissue samples and performed PCR experiments, which yielded results consistent with the findings from the TCGA liver cancer dataset (Fig. S2).
The KRS for each HCC patient was calculated using the following formula: KRS = (UAP1L1 × 0.0259) + (CENPA × 0.0336) + (TRIP13 × 0.0585) + (PLGLA × −0.0164) + (CDCA8 × 0.1891) + (PKIB × 0.0027) + (KIF20A × 0.0419) + (SLC16A3 × 0.0658). Based on this formula, HCC patients were stratified into high- and low-KRS groups, with the median KRS value of 0.578 serving as the threshold for categorization.
Evaluation and validation of kinase‐related gene signatureFigure 5A presents the relationship between KRS and survival duration for each patient within the TCGA-LIHC cohort. PCA successfully distinguished between the high- and low-KRS groups, as shown in Fig. 5B. Kaplan–Meier survival analysis further revealed that the high-KRS group had significantly inferior OS, DFI, and PFI in comparison to the low-KRS group (Fig. 5E–G). To assess the prognostic accuracy of KRS, we constructed a ROC curve, which yielded AUC values of 0.746, 0.715, and 0.691 for 1-year, 2-year, and 3-year survival, respectively (Fig. 5I). Additionally, the prognostic efficacy of KRS was corroborated in a separate cohort of 231 HCC samples from the ICGC database, utilizing the same prognostic model (Fig. 5C–D, H, J). Kaplan–Meier analysis of this ICGC validation cohort confirmed that the high-KRS group had a shorter OS compared to the low-KRS group (Fig. 5H). The AUC values for KRS in the ICGC cohort were 0.759, 0.734, and 0.747 for 1-year, 2-year, and 3-year survival, respectively (Fig. 5J). Collectively, these results highlight the effectiveness of our prognostic model, which is based on eight kinase-related genes, as a potent predictor of prognosis in HCC patients.
Fig. 5
Evaluation and validation of kinase‐related gene signature. Distribution of KRS and survival time, PCA analysis in TCGA-LIHC (A, B) and ICGC (C, D) cohorts. Kaplan–Meier curves of OS (E), DFI (F) and PFI (G) in TCGA-LIHC cohort. Kaplan–Meier curves of OS in ICGC cohort (H). ROC curves of 1-, 2-, and 3-year OS in TCGA-LIHC (I) and ICGC cohorts (J)
KRS is an independent prognostic predictor for HCC patientsThe results from univariate Cox regression analysis in the TCGA cohort revealed statistically significant correlations between KRS, T-stage, M-stage, and pTNM staging, and OS, as illustrated in Fig. 6A. Subsequent multivariate Cox regression analysis identified KRS as an independent prognostic factor for OS, exhibiting a HR of 4.667 (95% CI: 2.461–8.853) with a p-value of less than 0.001 (Fig. 6B). Similar findings were replicated in the ICGC cohort, further substantiating KRS's role as an independent prognostic factor (Fig. 6C, D).
Fig. 6
Predictive nomogram. Univariate and multivariate analysis of the clinicopathologic features and the KRS in TCGA-LIHC (A, B) and ICGC (C, D) cohorts. (E) Nomogram to predict the survival of the HCC patients. (F) Calibration curve for 1-, 3-, and 5-year OS. DCA (G) and ROC (H) curve of the clinicopathologic features and KRS
Development and validation of the nomogram prediction modelIn addition to our analyses, we integrated the collected clinicopathological characteristics with the kinase-related gene signature to construct a prognostic nomogram. This tool is designed for accurately forecasting 1-year, 3-year, and 5-year OS rates of HCC patients within the TCGA-LIHC cohort, as depicted in Fig. 6E. To validate its predictive accuracy, calibration curves for these OS intervals were compared against the ideal reference line. The close alignment observed in these comparisons, illustrated in Fig. 6F, underscores the nomogram’s robust predictive performance for the TCGA-LIHC cohort. Decision curve analysis (DCA) further demonstrated the nomogram's superiority over other study predictors (Fig. 6G). The AUC values indicating high predictive accuracy for 5-year survival (Fig. 6H).
Biological features of low- and high-KRS groupsIn our study, we delved into the underlying hallmark pathways that might account for the prognostic disparities observed between the low- and high-KRS groups in hepatocellular carcinoma. The analysis indicated a distinct enrichment of metabolism-related pathways in the low-KRS group, a finding that is clearly depicted in Fig. 7A. Conversely, the high-KRS group was characterized by a more pronounced involvement in cell cycle and cancer-related pathways, as detailed in Fig. 7B. This dichotomy in pathway activation underscores the potential mechanistic differences driving the varied prognoses associated with each KRS group.
Fig. 7
Biological features of low- and high-KRS groups. The GSEA pathway enrichment analysis in low- (A) and high- (B) KRS groups
KRS to assess tumor immune characteristicTo investigate the relationship between KRS and immune cell infiltration in HCC patients, we employed multiple analytical approaches to quantify the infiltration of diverse immune cells. This included conducting Spearman correlation analysis to elucidate the association between immune cell infiltration levels and KRS (Fig. 8A). Our analysis revealed a predominantly positive correlation between KRS and the infiltration of most immune cell types. This suggests that HCC patients with elevated KRS scores are likely to exhibit increased immune cell infiltration. Contrastingly, stroma scores demonstrated an inverse correlation with KRS. Utilizing the TIMER algorithm, we further delineated the disparities in immune cell infiltration between patients with high and low KRS scores. Notably, the high KRS group manifested significantly greater infiltration of B cells, neutrophils, macrophages, CD4+ T cells, and myeloid dendritic cells (Fig. 8B). Additionally, this group showed augmented expression levels of immune checkpoint genes (Fig. 8C), MSI scores (Fig. 8D), and MHC gene expression (Fig. 8E).
Fig. 8
KRS to assess tumor immune characteristic in HCC patients. The association between KRS and the multiple immune cell infiltration levels in TCGA-LIHC cohort (A). Boxplot for immune cell infiltration level (B) and immune checkpoint genes (C) between the high- and low-KRS groups. Violin and scatter plot for MSI scores between the high- and low- KRS groups (D). (E) Split violin plot for MHC genes between the high- and low-KRS groups
KRS to predict immunotherapy and chemotherapy responsesThe analysis of the IMvigor210 cohort showed that patients with a higher KRS tended to respond better to anti-PD-L1 immunotherapy (Fig. 9A). On the other hand, in the GSE78220 cohort with anti-PD-1 immunotherapy, the low KRS group exhibited a significant clinical response compared to the high KRS group (Fig. 9B). We also evaluated the ability of the KRS to predict drug sensitivity for conventional chemotherapy drugs. Our findings revealed that the high KRS group had significantly lower IC50 values for all the tested drugs (Fig. 9C).
Fig. 9
The role of the KRS in predicting treatment response. (A) Boxplot for the KRS between anti-PD-L1 immunotherapy response and non-response groups in the IMvigor210 cohort (blue, response; red, non-response). (B) Boxplot for the KRS between anti-PD-1 immunotherapy response and non-response groups in the GSE78220 (blue, response; red, non-response). (C) Boxplot for the the half maximal inhibitory concentration (IC50) values of the chemotherapy responses between low and high KRS patients
Kinase hub gene verificationTo investigate the impact of genes associated with kinases on tumors, we performed tissue validation on the core genes within the aforementioned model. Firstly, subsequent to thorough analysis, we identified five genes (CDK4, PLK1, AURKA, AURKB, BUBIB) that hold significant prominence among the kinase-related genes (Fig. S3). We commenced by validating the differential expression of these five genes in tumors in comparison to the adjacent normal tissues. Our analysis revealed expression disparities in PLK1, AURKA, and AURKB, as illustrated in Fig. 10A. Moreover, we scrutinized the connection between these five genes and clinical phenotypes. Our analysis divulged that liver cirrhosis bears associations with AURKB, CDK4, and PLK1. Notably, AURKB experiences downregulation while CDK4 undergoes upregulation in instances of liver cirrhosis, as depicted in Fig. 10B. Additionally, regarding tumor necrosis, we observed elevated expressions of AURKB and PLK1 in the necrotic group, as opposed to the non-necrotic group, as illustrated in Fig. 10C. Finally, to gauge the potential impact on liver cancer prognosis, we conducted survival analysis on the five genes. Intriguingly, excluding PLK1, the remaining four genes demonstrated prognostic significance for liver cancer, as depicted in Fig. 10D. Based on the clinical phenotypes of each patient and the differential expression of different core genes, we performed univariate and multivariate COX regression to identify independent prognostic factors for liver cancer. Our analysis revealed that in addition to the four core genes, age, HCV infection prognosis, and bile duct invasion were independent risk factors affecting the prognosis of liver cancer patients (Supplementary Table 7). Therefore, based on our analysis of these independent factors, we constructed a nomogram graph for clinical convenience (Fig. 10E).
Fig. 10
Core gene expression validation. (A) Differential expression of five core genes in liver cancer. (B) Association between core genes and liver cirrhosis. (C) Core genes associated with tumor necrosis. (D) Core genes impacting prognosis of liver cancer. (E) nomogram for the Core gene model
Comments (0)