
Remember me
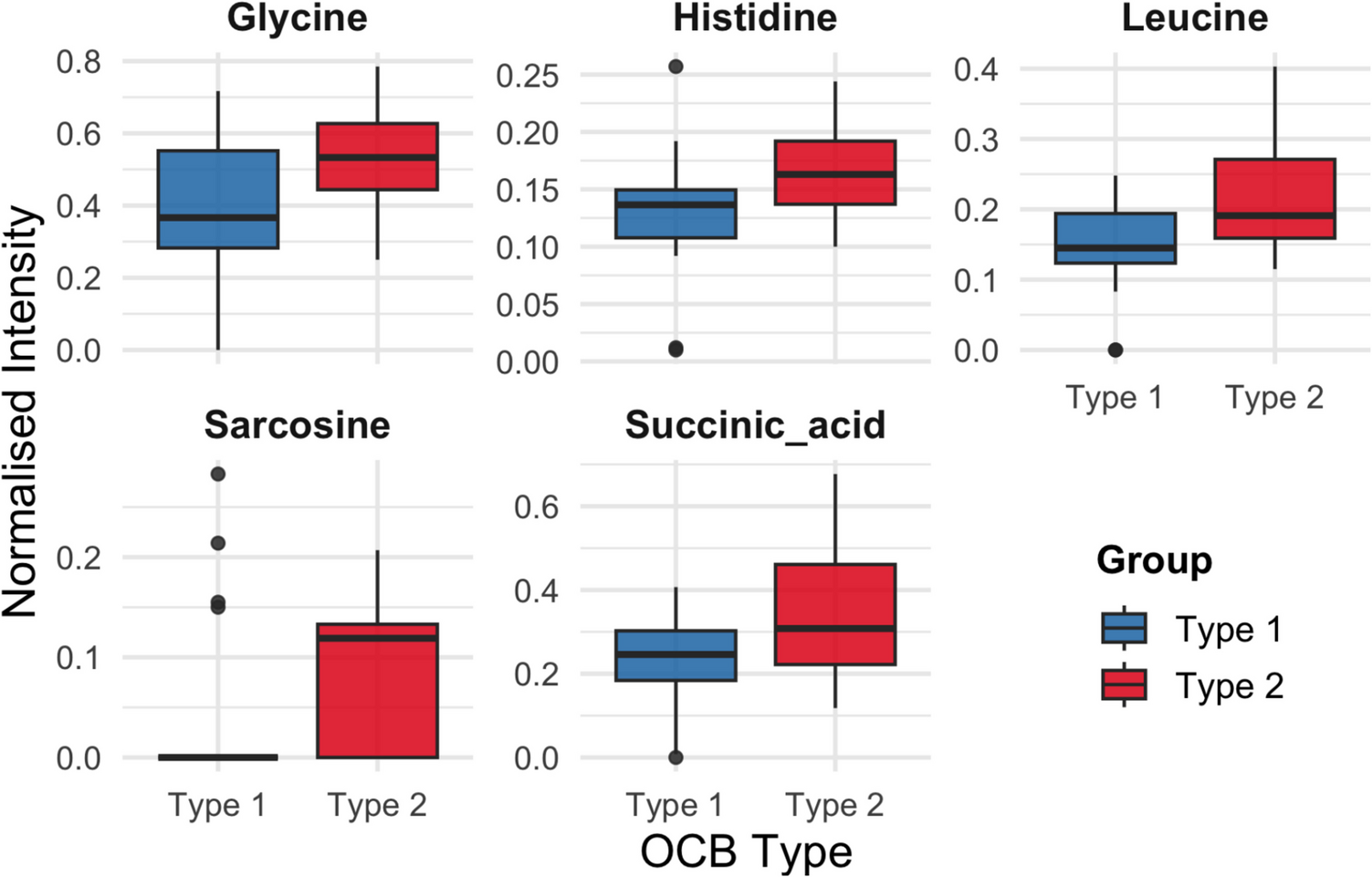


Mutant ATXN1[82Q] expression alters mitochondrial protein expression in mouse cerebellar tissue. Our lab and others (Ripolone et al. 2018; Ferro et al. 2017b; Stucki et al. 2016) previously demonstrated mitochondrial OXPHOS deficits in cerebellar tissue of SCA1 mice models preceded disease pathology and symptom onset. In order to gauge whether functional mitochondrial genes apart from the electron transport genes are altered in SCA1 cerebellum, we conducted a meta-analysis by cross-referencing the Ingram et al. (Ingram et al. 2016) SCA1 RNAseq dataset with the MitoCarta 2.0 database. The Ingram dataset contained genes from whole cerebellar tissue of 5-, 12-, and 28-week FVB/n wild type (WT) mice, ATXN1[82Q-S776] (Progressive SCA1, B05 line) mice, and ATXN1[30Q-D776] (Non-progressive SCA1) mice. At the time of our meta-analysis, the MitoCarta2.0 database consisted of 1158 mouse genes experimentally determined to exhibit mitochondrial function. A subfraction of 27–32 genes showed differential expression in the Progressive or Non-progressive SCA1 cerebellar tissue compared to WT cerebellar tissue, with a total of 40 genes (3.5%) showing differential expression at one or more developmental ages (Fig. 1A).
Fig. 1
Mutant ATXN1 expression in cerebellar Purkinje neurons selectively alters nuclear-encoded mitochondrial gene expression and oxidative stress in cerebellar tissue. A-B) Datasets displaying genes with significant differential expression between Progressive SCA1 and WT, and between Non-progressive SCA1 and WT in cerebellar tissue were re-analyzed. Graphed are differential expression ratios of MitoCarta2.0/Ingram et al. genes in cerebellar tissue from ATXN1[30Q-D776] (Non-progressive SCA1) mice compared to WT mice A and ATXN1[82Q-S776] (Progressive SCA1, B05 line) mice compared to WT mice B at 5, 12 and 28 weeks of age (x-axis). (C-E) SCA1 mice cerebellar tissue displays selective forms of oxidative stress; specifically, increased protein carbonylation C, but not DNA oxidative damage D or reducing ability E. *P < 0.05
The most obvious feature of our meta-analysis is the increased expression of genes in the Progressive or Non-progressive SCA1 tissue compared to WT, with only a small number of genes showing decreased expression. Among the latter are Casp3 (NCBI Ref Seq NM_001284409.1; caspase-3), the execution-phase apoptotic cysteine-aspartic acid protease, which shows greater downregulation in Progressive SCA1 than Non-progressive SCA1. Aldh9a1 (NM_019993.4; aldehyde dehydrogenase 9, subfamily A1), an electron acceptor enabling oxidoreductase activity, shows similar and slight downregulation in both the Progressive and Non-progressive SCA1 compared to WT at 12 weeks of age.
The sodium/glucose co-transporter gene Slc5a1 (NM_019810.4) and centrosomal proteins Cep72 (NM_028959) and Cep76 (NM_001081073) show elevated expression levels in Non-progressive SCA1 mouse cerebellum compared to WT, with the highest expression apparent at 12 weeks. In contrast, the transmembrane protein Tmem255a (NM_172930) shows elevated, but decreasing expression with age, while the hexokinase Hk2 (NM_013820) shows elevated, but increasing expression with age (Fig. 1A).
The guanine nucleotide binding protein Gng13 (NM_022422), Fam107b (NM_025626), sodium/proton exchanger Slc9a3 (NM_001081060), and cytochrome c oxidase subunit Cox6b2 (NM_183405) show greater expression in Progressive SCA1 mouse cerebellum compared to WT. Of those, Gng13 and Slc9a3 increase with age, Fam107b shows highest levels of expression at 12 weeks, and Cox6b2 decreases with age (Fig. 1B). Overall, the results of our meta-analysis show substantial differential regulation of expression of multiple functional mitochondrial genes, beyond just those involved in oxidative phosphorylation.
ATXN1[82Q] expression in SCA1 mice cerebellum produces features of selective oxidative stress. Previously, we identified oxidative phosphorylation chain complex I dysfunction in whole cerebellar tissue from B05 mice (Ferro et al. 2017b). We now verify that selective measures of oxidative stress are increased compared to WT cerebellar tissue. Protein carbonylation, a form of protein oxidation promoted by ROS generation, is significantly elevated in SCA1 (3.849 ± 0.3422 nmol per mg tissue) compared to WT (2.902 ± 0.2104 per mg tissue) (p < 0.05) (Fig. 1C). However, no difference in oxidative DNA damage (B05: 219.5 ng ± 15.68 ng per mg genomic DNA; WT: 227.0 ± 79.00 ng per mg genomic DNA; P = 0.8958) (Fig. 1D) or oxidative capacity (B05: 0.2524 ± 0.0022 mmol FeSO4; WT: 0.2491 ± 0.0079 mmol FeSO4; P = 0.6934) (Fig. 1E) was detected. Taken together, these results indicate that expression of ATXN1[82Q] protein in cerebellar Purkinje neurons produces select cytoplasmic metabolic deficits.
ATXN1[82Q] expression produces cytoplasmic physiological deficits in SCA1 cell models. We next assessed whether expression of the ATXN1[82Q] transgene in cerebellar-derived cell models of SCA1 correlates with cytoplasmic deficits. We used a Daoy line, a human medulloblastoma cell which endogenously expresses ATXN1, in which RFP-ATXN1[82Q] is stably expressed (Park et al. 2013; Huang et al. 2022; Lagalwar 2022) (SCA1 cells). First, we measured ATP levels in WT (100.0 ± 7.284%) and SCA1 cells (25.73 ± 1.598%) under resting conditions and found that SCA1 cells featured a statistically decreased amount of ATP production (P < 0.0001) (Fig. 2A). Oxidative stress in the form of the cell permeable ROS H2O2, under resting conditions, was measured next. WT cells exhibited a non-significant trend of reduced H2O2 production (by 75.92 ± 7.622%) compared to SCA1 cells (Fig. 2B). Whole cell calcium activity of SCA1 cells compared to WT cells (Fig. 2C-E) was significantly increased (P < 0.0001). Moreover, application of 1 μM STR for 3 h evoked a strong calcium response in SCA1 cells (Fig. 2G) compared to WT cells (Fig. 2F), exhibited by increased signal intensity (Fig. 2G). Taken together, these results show that under resting conditions, expression of ATXN1[82Q] produces cytoplasmic physiological deficits and suggest that Daoy cells may be a good model for studying those deficits in the context of SCA1.
Fig. 2
Mutant ATXN1 expression in cerebellar-derived Daoy cells alters cellular physiology. A During rest conditions, SCA1 cell models expressing RFP-ATXN1[82Q] and endogenous ATXN1 display decreased ATP production compared to WT Daoy cells. B During rest conditions, SCA1 cell models display increased H2O2 ROS production compared to WT Daoy cells. C-E During rest conditions, SCA1 cell models D display increased GFP fluorescence of the calcium indicator dye Fluo-4AM compared to WT Daoy cells E. Following 3 h of cellular stress instigation with 1 μM STR F-G, SCA1 cells G selectively feature enhanced Ca.2+ granule formation, compared to WT cells F. *** P < 0.001
ATXN1[82Q] expression produces mitochondrial morphological and localization deficits in SCA1 cells. In order to determine whether ATXN1[82Q] expression affects mitochondrial morphology, we visualized their ultrastructure in WT and SCA1 cells (Fig. 3A-D). WT mitochondria appear oval-shaped with intact outer membrane and cristae stretched through the organelle (Fig. 3A). SCA1 mitochondria, in contrast, feature rounded morphology, vacuoles and disrupted cristae (Fig. 3B-D). To visualize alterations in mitochondrial localization, we labeled the MICOS complex Mic60 subunit using Mitofilin (green) (Fig. 3E). RFP-ATXN1[82Q] and endogenous ATXN1 were labeled with the 11NQ polyclonal antibody (red). Large aggregates of RFP-ATXN1[82Q]/ATXN1 appear primarily nuclear (DAPI). However, smaller aggregates and more diffuse protein appear cytoplasmic. Notably, when nuclear aggregates are present, mitochondria display perinuclear localization (Fig. 3E). Next, RFP-ATXN1[82Q-A776] and endogenous ATXN1 were labeled with 11NQ antibody (red), Mic60 (green), and DAPI (blue) (Fig. 3F). In contrast to SCA1 cells (Fig. 3E), ATXN1[82Q-A776] cells retained diffuse nuclear ATXN1, likely endogenous, and diffuse mitochondrial Mic60 throughout the cytoplasm. These results indicate gross, widespread mitochondrial changes in SCA1 cells caused by expression of the disease-prone ATXN1[82Q] protein.
Fig. 3
Mutant ATXN1 expression in cerebellar-derived Daoy cells disrupts mitochondrial morphology and localization. A-D Transmission electron micrograph (TEM) imaging of mitochondria in WT cells A and SCA1 cells B-D. SCA1 mitochondria are distorted with cristae disruptions, large vacuoles and a rounded morphology. E RFP-ATXN1[82Q] (red) stained for Mitofilin/Mic-60 (green) display perinuclear mitochondrial localization when ATXN1 nuclear aggregates are present. F RFP-ATXN1[82Q-A776] (red) stained for Mitofilin/Mic-60 (green), display nuclear diffuse ATXN1 and cytoplasmic mitochondrial localization
ATXN1[82Q] expression alters mitochondrial protein production in SCA1 cells. To assess whether mitochondrial protein levels are affected by expression of RFP-ATXN1[82Q], we compared the inner mitochondrial membrane protein Optic Atrophy 1 (OPA1), the mitochondrial fusion regulator Mitofusion2 (MFN2), and the mitochondrial import protein TOM20 in the three cell lines. Compared to WT cells (Fig. 4A) or ATXN1[82Q-A776]-expressing cells (Fig. 4B), OPA1 expression (green) displays decreased diffuse, cytoplasmic expression in SCA1 cells and instead localizes perinuclearly, as shown against the blue nuclear DAPI stain (Fig. 4C-D).
Fig. 4
Mutant ATXN1 expression in cerebellar-derived Daoy cells alters mitochondrial protein expression. A-D Cytoplasmic dynamin-related GTPase OPA1 (green) displays greater levels of cytoplasmic expression in WT A and RFP-ATXN1[82Q-A776] B cells than in SCA1 cells C-D. DAPI nuclear staining is shown in blue. (E) Protein expression of mitofusin 2 (MFN2) and TOM20 is decreased in cells expressing ATXN1[82Q] transgenes compared to WT cells. * P < 0.05, **** P < 0.0001
In contrast, protein expression analysis of MFN2 and TOM20 reveals decreased levels of both proteins in the transgenic lines compared to WT cells (Fig. 4E). Specifically, one-way ANOVA of MFN2 expression in the three cell lines [F (2, 177) = 12.67, p < 0.0001] shows no significant difference in expression levels between SCA1 and ATXN1[82Q-A776] cells (normalized mean value of 73.34 and 75.11, respectively; Tukey’s post hoc analysis p = 0.9521). However, significant decreases in expression are found between SCA1 and WT cells (normalized mean value of 73.34 and 100.00, respectively; Tukey’s post hoc analysis p < 0.0001) and between ATXN1[82Q-A776] and WT cells (normalized mean value of 75.11 and 100.00, respectively; Tukey’s post hoc analysis p = 0.0001).
One-way ANOVA of TOM20 [F (2, 237) = 14.62, p < 0.0001] shows no significant difference in expression between SCA1 and ATXN1[82Q-A776] cells (normalized mean value of 70.67 and 68.50, respectively; Tukey’s post hoc analysis p = 0.9216). Similarly, significant decreases in expression are found between SCA1 and WT cells (normalized mean value of 70.67 to 100.00, respectively; Tukey’s post hoc analysis p < 0.0001) and between ATXN1[82Q-A776] and WT cells (normalized mean value of 68.50 to 100.00, respectively; Tukey’s post hoc analysis p < 0.0001).
The results of Fig. 4 indicate differential regulation of mitochondrial proteins in the presence of ATXN1[82Q] and ATXN1[82Q-A776]. While OPA1 expression is selectively altered in SCA1 cells compared to WT and ATXN1[82Q-A776], MFN2 and TOM20 are equally affected in the presence of both transgenic ATXN1 polyglutamine-expanded proteins, regardless of their phosphorylation status. Importantly, the selective alteration of mitochondrial gene expression by ATXN1[82Q-A776] in particular, which cannot be phosphorylated at the 776 residue and therefore is not readily nuclear-translocated, indicates that it is cytoplasmic ATXN1 that is causing the disruption.
ATXN1[82Q] expression alters mitochondrial physiology in SCA1 cell models. We next assessed energy metabolism and oxidative phosphorylation. SCA1 cells display higher extracellular acidification rates (ECAR, a measure of cellular glycolysis) and higher oxygen consumption rates (OCR, a measure of mitochondrial oxidative phosphorylation) than WT cells, pushing them into an “energetic” phenotype indicative of cellular stress. Interestingly, cells stably expressing ATXN1[82Q-A776] display a phenotype in-between the WT and SCA1 models (Fig. 5A). Notably, ATXN1[82Q-A776] cells also display intermediate levels of basal respiration (Fig. 5B), proton leak (Fig. 5C) and coupling efficiency (Fig. 5D). The low coupling efficiency of SCA1 cells, which is a measure of the percentage of respiration used for ATP synthesis, appears in line with low ATP levels (Fig. 2A).
Fig. 5
Mutant ATXN1 expression in cerebellar-derived Daoy cells alters mitochondrial physiology. A WT cells display low bioenergetics, measured by the Seahorse MitoStress test, indicative of a quiescent state compared to the high bioenergetics displayed by SCA1 cells, indicative of an energetic, stressed state. In contrast, ATXN1[82Q-A776] cells display intermediate extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) phenotypes C, indicative of an in-between state. The energy map data is reflective of the SCA1 cells having higher basal respiration levels B, enhanced proton leak C and decreased ATP production coupling efficiency D than WT cells, with ATXN1[82Q-A776] cells again displaying intermediate phenotypes. E Red channel and green channel mitochondrial membrane potential emission from individual WT cells, SCA1 cells and ATXN1[82Q-A776] cells following loading with JC-10 dye and masking solution was imaged and quantified. Of note is the inclusion of RFP-tagged-ATXN1[82Q] and RFP-tagged-ATXN1[82Q-A776] in the red channel emission. Cells were next treated with 1 μM STR, imaged after three hours, and their green channel emission were plotted. F-G Time-lapse imaging following specific JC-10-loaded ATXN1[82Q] cells over eighty minutes of STR treatment shows a gradual decrease in red channel emission and a gradual increase in green channel emission, indicative of depolarizing mitochondrial membrane potential. F Red and green emission traces from a single RFP-ATXN1[82Q] cell (shown with white circle) over time following STR treatment. **** P < 0.0001
Finally, we assessed mitochondrial membrane potential (MMP) by its green:red ratio, a measure of overall mitochondrial health. Importantly, RFP levels in the SCA1 cells (mean intensity = 17.33) were lower than the 540 nm red channel emittance (mean intensity = 236.9) and did not affect the MMP signal of SCA1 cells. Under resting conditions, WT, SCA1 and ATXN1[82Q-A776] cells emitted statistically similar levels of red (mean intensities = 241.3, 236.9, 242, respectively; [F (2, 84) = 1.766, p = 0.1773]) and green (mean intensities = 118.7, 98.25, 95.24, respectively; [F (2, 64) = 2.324, p = 0.1061] fluorescence (Fig. 5E).
We next measured the response to STR-induced cellular stress. Treated SCA1 cells featured increased green wavelength emission (mean intensity = 144.3) compared to treated WT (mean intensity = 43.21) or treated ATXN1[82Q-A776] cells (mean intensity = 56.10). One-way ANOVA analysis confirmed the statistical increase in the green MMP emission from treated SCA1 cells [F (2, 104) = 55.67, p < 0.0001] (Fig. 5E, 3 hr STR). Tukey’s post hoc analysis p-values between WT and SCA1 cells (p < 0.0001), between SCA1 and ATXN1[82Q-A776] cells (p < 0.0001), and between WT and ATXN1[82Q-A776] cells further confirm that only the SCA1 line displayed mitochondrial vulnerability to cellular stress.
In order to visualize the change in SCA1 cells during stress induction, we used time-lapse imaging to calculate maximum emission from an individual whole z-stacked SCA1 cell (white circle) over 80 min post-induction (Fig. 5F). The graph in Fig. 5F shows decreased red channel emission and the corresponding increased green channel emission during that time period (Fig. 5F). To visualize population changes, field images were taken at 540 nm and 490 nm every 10 min (Fig. 5G). The visual reduction in red signal and corresponding increase in green signal can be seen in Fig. 5G.
The results of Fig. 5 support that mitochondrial physiology is altered in the presence of mutant ATXN1. Taken together, the results of Figs. 2, 3, 4 and 5 indicate that widespread cytoplasmic and mitochondrial morphological, biochemical and physiological changes occur in SCA1 cell models in the presence of mutant ATXN1[82Q] expression.
Does cytoplasmic ATXN1[82Q] directly contribute to mitochondrial dysfunction? We addressed this question in the following ways. First, we investigated whether mitochondrial-targeted compounds alter the physiological deficits induced by the expression of ATXN1[82Q]. Increased ROS H2O2 is increased in SCA1 cells compared to WT cells at rest (Fig. 2B). Moreover, the electron transport chain complex II substrate, succinic acid, significantly decreased H2O2 ROS levels (67.1667 ± 7.640%); p < 0.05) (Fig. 6A). Increases in concentrations up to 3.4 mM resulted in greater decreases in H2O2 ROS levels (0.034 mM: 57.576 ± 0.599%; 0.34 mM: 58.170 ± 3.400%; 3.4 mM: 57.990 ± 1.515%; all: p < 0.01) (Fig. 6A).
Fig. 6
Mitochondrial-targeted compounds alter the cellular and mitochondrial physiological deficits of SCA1 cells. A Treatment of SCA1 cells with OXPHOS complex II substrate succinic acid reduces H2O2 ROS production. B Under high (25 mM) glucose conditions, H2O2 ROS production in SCA1 cells can be significantly decreased with the addition of the mitochondrial-targeted antioxidant, MitoQ. Under high (10 mM) galactose conditions, MitoQ increases H2O2 ROS production in SCA1 cells. (C-H) Green to red channel emission ratio of vehicle-treated (Veh) WT cells and SCA1 cells, and SCA1 cells treated with the OXPHOS complex I inhibitor rotenone (Rote), succinic acid (SA) or both, was imaged and quantified. Representative green/red overlay images C are shown in D-H. * P < 0.05, *** P < 0.001, **** P < 0.0001
We next measured the production of ROS H2O2 in SCA1 cells in the presence of the mitochondrial-selective antioxidant, MitoQ (Stucki et al. 2016), under either high (25 mM) glucose conditions or high (10 mM) galactose conditions (Fig. 6B). MitoQ reduced H2O2 production in SCA1 cells (68.67 ± 11.76%; P < 0.05) (Fig. 6B) compared to WT levels under high glucose conditions. Under high galactose conditions, 500 nM MitoQ increased H2O2 production in SCA1 cells (132.3 ± 4.133%) compared to WT cells (100.0 ± 3.783%; P < 0.001) (Fig. 6B), perhaps due to compensation in the context of enhanced elimination during high metabolism.
Finally, we measured mitochondrial membrane potential in vehicle-treated WT and SCA1 cells, and SCA1 cells treated with the complex I inhibitor rotenone, succinic acid, or both [F (4, 627) = 35.02, p < 0.0001]. The MMP green:red ratio of SCA1 cells (n = 180, mean ratio = 0.4732 ± 0.03; Fig. 6C, 6E) to WT cells (n = 90, mean ratio = 0.2290 ± 0.01; Fig. 6C-D) was calculated. Significant differences between the SCA1 to WT ratios were determined by post hoc analysis (p < 0.0001). Treatment of SCA1 cells with 600 nM rotenone for 3 h (n = 122, mean ratio = 0.6845 ± 0.03; Fig. 6C, 6 F) caused an increase in the MMP ratio compared to vehicle treatment (post hoc analysis of p < 0.0001). In contrast, the overnight addition of 3.4 μM succinic acid to SCA1 cells (n = 120, mean ratio = 0.3911 ± 0.03; Fig. 6C, 6G) did not significantly alter the MMP ratio (p = 0.1108). Overnight pre-treatment with 3.4 μM succinic acid prior to adding rotenone (n = 120, mean ratio = 0.4386 ± 0.02; Fig. 6C, 6H) significantly reduced the MMP ratio compared to rotenone-treatment alone (p < 0.0001). No significant difference was calculated between succinic acid/rotenone-treated SCA1 cells and succinic acid-treated SCA1 cells (p = 0.7048).
The results of Fig. 6 suggest that alteration of mitochondria by enhancing electron transport chain function with succinic acid, or neutralizing mitochondrial reactive oxygen species with MitoQ, can mitigate the enhanced oxidative stress and mitochondrial membrane potential seen in SCA1 cells. Conversely, inhibition of electron transport chain function with rotenone exacerbates mitochondrial membrane potential in the presence of ATXN1[82Q].
To assess direct interactions of mutant ATXN1 with cytoplasmic and mitochondrial proteins, we first conducted a meta-analysis of previously published data. Zhang, Williamson and Bogoyevitch (Zhang et al. 2018) developed complementary proteomics strategies (biotinylation, GFP-trap pulldown) to identify proximal (via GFP-trap pulldown) and direct (via biotinylation and GFP-trap pulldown) interactome partners of mutant ATXN1[85Q] in neuro2A cells. Both strategies were conducted under resting and oxidative stress conditions, the latter of which were induced by arsenate. We filtered their shared dataset through DAVID and MitoCarta 2.0 (Fig. 7). Three apoptotic proteins, 8 OXPHOS proteins, 8 glycolytic proteins and the mitochondrial transcription Mterf1b were identified as potential interactors of ATXN1[85Q-S776] by one of their four strategies/conditions. A combination of two of the strategies/conditions identified an additional 3 apoptotic proteins, 3 OXPHOS proteins, 3 mitochondrial membrane proteins, and 7 glycolytic proteins as potential interactors. A combination of three of the strategies/conditions identified an additional two apoptotic proteins, the mitochondrial membrane protein Slc25a12, and the glycolytic protein Galk1 as potential interactors. Finally, the mitochondrial membrane protein Slc25a31, which when mutated causes “Combined Oxidative Phosphorylation Deficiency 34” was identified as a potential interactor of ATXN1[85Q-S776] by all four proteomic strategies/conditions.
Fig. 7
Mutant ATXN1 interacts with and/or proximally localizes to a host of cytoplasmic and mitochondrial proteins. Filtered analysis of interactomic data from Zhang et al. 2018 (Burright et al. 1995) represents cytoplasmic and mitochondrial proteins interacting with and/or in proximity of mycBioID-ATXN1[85Q] or GFP-Trap-ATXN1[85Q] in N2a cells. Mitochondrial and glycolytic proteins that bound ATXN1[85Q] were identified through Mouse MitoCarta 2.0 cross-indexing and DAVID Functional Annotation Bioinformatics Analysis (https://david.ncicrf.gov). Proximal localization and direct interactions are shown under rest, stressed and both rest and stressed conditions. *Purkinje neuron-enriched gene
To further analyze the data, we highlighted, in blue, the combined biotinylation and GFP-trap pulldown data for cells at rest (Fig. 7). Next, we highlighted, in yellow, the combined biotinylation and GFP-trap pulldown data from the stressed condition experiments (Fig. 7). Proteins that were identified in both conditions were highlighted in green (Fig. 7). A small subset of proteins were only identified under stressed conditions indicating that the vast majority of glycolytic and mitochondrial proteins interact with ATXN1[85Q] under normal conditions.
We also investigated co-localization from z-stack slices of Mic60 and ATXN1 in WT, SCA1 and ATXN1[82Q-A776] cells. Figure 8 shows slices from the top, middle and bottom of 20 μm z-stacks of WT (A-C), SCA1 (D-F) and ATXN1[82Q-A776] cells (G-I) stained with 11NQ, Mic60 and DAPI. In all three cell types, co-localization is visible in the middle z-layers (B,E,H). However, the degree of co-localization is reduced in WT cells (A-C), compared to SCA1 cells (D-F) and ATXN1[82Q-A776] cells (G-I). Figure 8J displays an orthogonal rotation of Fig. 8H depicting co-localization in the middle of the cell. Overall, the interactomic data and co-localization analysis indicates that mutant ATXN1 interacts proximally with cytoplasmic proteins.
Fig. 8
Z-stack imaging of WT, SCA1 and ATXN1[82Q-A776] cells capture co-localization between the ATXN1[82Q] transgenic proteins and MICOS complex Mic60 mitochondrial protein. (A-C) Endogenous ATXN1 (red) in WT cells does not co-localize with Mic60 (green) in z-stack layers. In contrast, ATXN1[82Q] (D-F, red) and ATXN1[82Q-A776] (G-I, red) extensively co-localize (yellow) with Mic60 (green), particularly within the middle layers (B, E, H) of a 20 μm z-stack. (J) Co-localization can be seen in yellow from the rotation of the z-stack depicted in E
Comments (0)