The c.494del p(Pro165Glnfs*9) mutation has been reported in only two prior studies, but the phenotype remains poorly characterized [2, 7]. Oliviera et al. described one patient with an identical variant who presented with splenomegaly, portal vein thrombosis, headache, Hgb 17.1 g/dL, and EPO 51 mlU/mL [2]. However, clinical courses, management, and outcomes are unknown. In Dewey et al., of 22 individuals with unspecified frameshift mutations in Pro165 of EGLN1, only seven patients had erythrocytosis, and there is no further discussion. Mutations in PHD2 have also been implicated in cases of paragangliomas indicating tumor suppressor activity, but it remains unclear whether patients with ECYT3 have an increased risk for these tumors [8, 9].
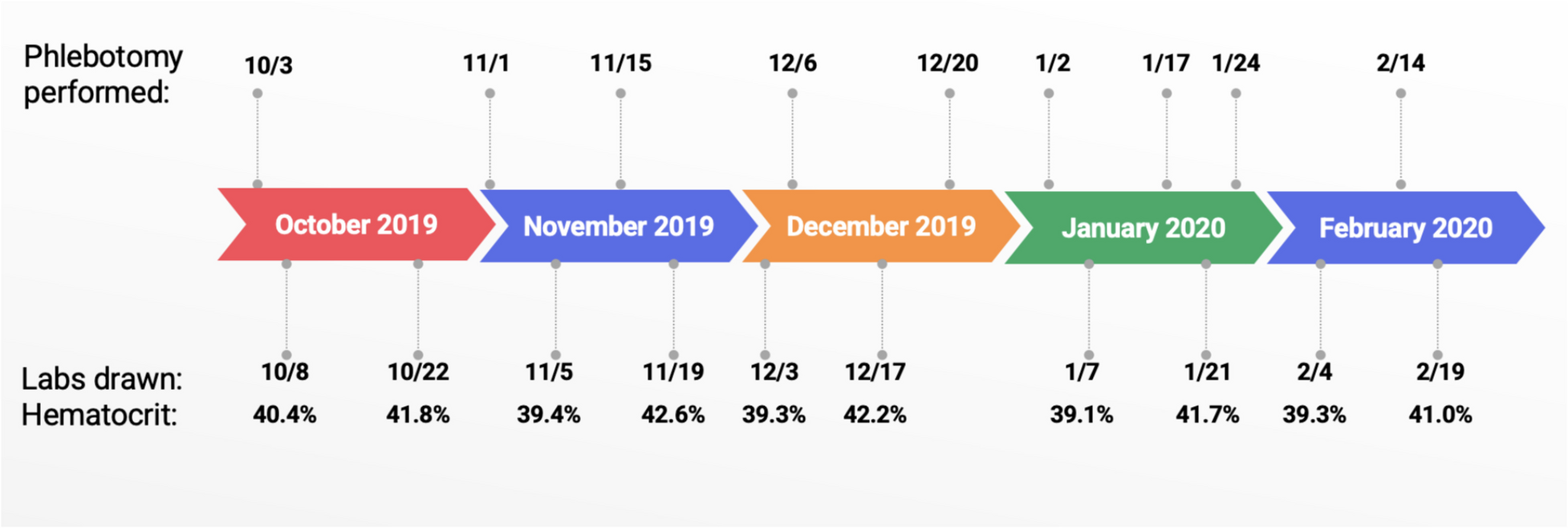
Although this patient’s presentation is largely similar to other variants of ECYT3, several key features set this case apart. The patient’s severe and refractory vasomotor symptom burden was out of proportion to the aggressive hematocrit target of 38–40%. Symptom control required a delicate balance between avoiding erythrocytosis symptoms and inducing anemia. Some symptoms, namely pruritus and headache, could not be entirely controlled by routine venesection and have been unresponsive to alternative medications.
His medical history has since been complicated by multiple thrombotic events, raising concerns for an underlying hypercoagulable state. He experienced a retinal artery occlusion, venous insufficiency, and thrombophlebitis following superficial vein thrombosis. Taken together, these incidents combined with the patient’s persistent erythrocytosis suggest a predisposition to thrombophilia in ECYT3.
This patient’s EPO was normal at the time of testing, a counterintuitive finding given PHD2’s role in suppressing EPO production. Similar findings were reported in other cases of ECYT3, though none with this particular variant [10]. The only other documented case of this variant with laboratory data had an abnormally high erythropoietin [2]. One hypothesis is that erythrocytosis with low to normal EPO could point towards increased sensitivity or expression of the EPO receptor [10]. A second theory is that ECYT3 creates a new HCT “set-point,” causing an elevation of EPO after phlebotomy, returning to normal once the patient reaches that set point. This phenomenon was observed in a subset of patients with familial erythrocytosis type 2 (ECYT2), defined by loss of function of VHL [3, 11, 12].
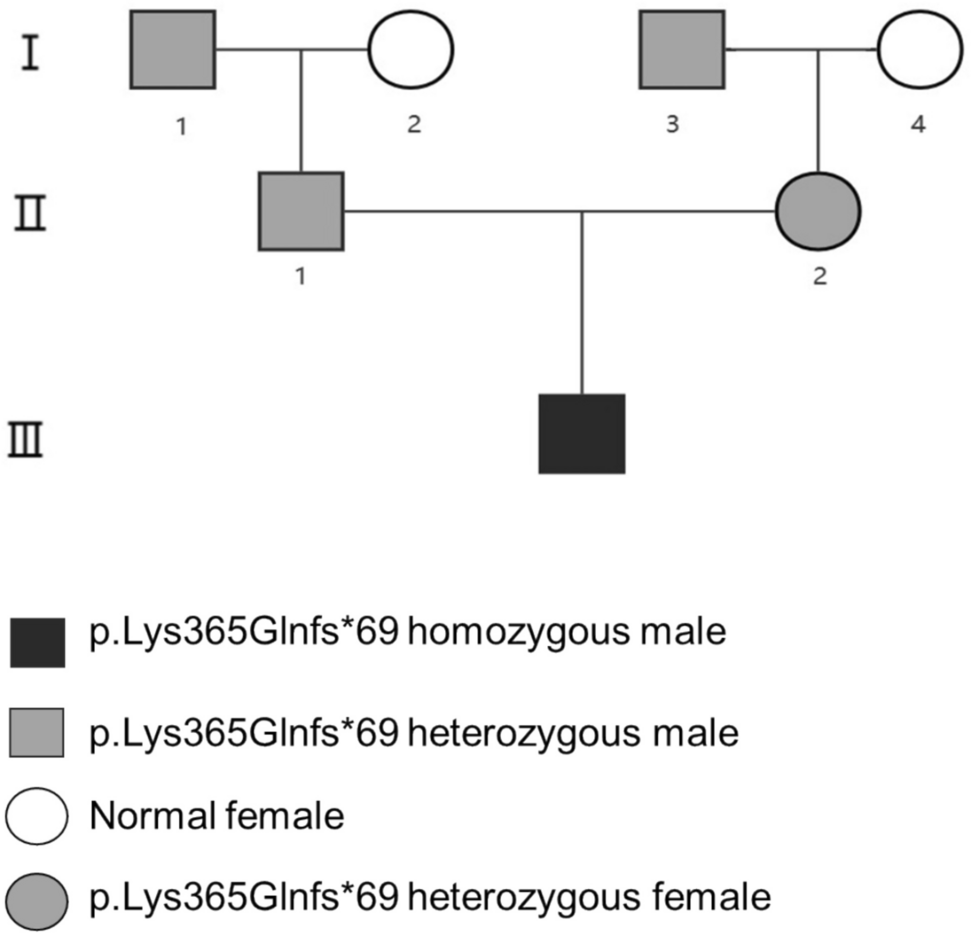
This case study illustrates the complexity of ECYT3 through a comprehensive patient journey spanning nearly two decades. Our patient has provided a detailed family and personal medical history and documentation of his symptoms. However, there are several limitations, including a lack of familial data. Anecdotally, his son had the same variant of PHD2 with similar symptoms, though his health information is unknown. More clinical data and further studies of patients with ECYT3 are necessary to understand the pathophysiology and ideal treatment, particularly with the c.494del p(Pro165Glnfs*9) variant. To improve our understanding of ECTY3 and identify appropriate treatments, we urge providers to test patients with unexplained erythrocytosis for germline genetic mutations. Clinical findings in patients harboring germline variants in PHD2 should be reported to build a stronger evidence base and inform future studies on ECYT3.




Comments (0)