
Remember me





Human brain samples for this study were obtained from donors with a clinical and neuropathological diagnosis of PD and controls not meeting clinical or neuropathological diagnostic criteria for any neurodegenerative disease. Specifically, the CN and Pu were dissected from coronal slabs at the level of the nucleus accumbens. The control group comprised 27 individuals (Pu = 27, CN = 24) ranging in age from 25 to over 90 years. The PD group included 36 individuals (Pu = 35, CN = 34) aged between 60 and over 90 years. Samples were collected from three sources: the Human Brain and Spinal Fluid Resource Center (Los Angeles, CA, USA), the Parkinson’s UK Brain Bank at Imperial College London (London, UK), and the Massachusetts Alzheimer’s Disease Research Center (Charlestown, MA, USA). Donors or their next-of-kin provided written informed consent for brain autopsy, and the study was approved by the review board of each brain bank. Detailed information regarding the samples is provided in Supplementary Table 1. Since PD is more common in men, the control and PD groups were not matched by sex (i.e., there were 19 females and 44 males in the full cohort, control: 18/9 and PD: 26/10 male/female). Consequently, the study lacks sufficient power to robustly analyze sex-specific effects, such as differences in cell class proportions or the influence of sex on gene expression regulation.
Tissue dissociationNuclei isolation from fresh frozen tissue was conducted following the protocol outlined by the Allen Institute for Brain Science (https://www.protocols.io/view/isolation-of-nuclei-from-adult-human-brain-tissue-eq2lyd1nqlx9/v2), with specific guidelines. All procedures were performed at 4 °C to maintain sample integrity. Tissue samples (100–150 mg) were thawed on ice and homogenized in 2 mL of chilled, nuclease-free homogenization buffer. The buffer composition included 10 mM Tris (pH 8), 250 mM sucrose, 25 mM KCl, 5 mM MgCl2, 0.1 mM DTT, protease inhibitor cocktail (1 × , 50 × stock in 100% ethanol, G6521, Promega), 0.2 U/μL RNasin Plus (N2615, Promega), and 0.1% Triton X-100. Homogenization was carried out using a dounce tissue grinder with loose and tight pestles (20 strokes each; 357,538, Wheaton). The nuclei solution was filtered sequentially through 70 μm and 30 μm strainers. Tubes and strainers were rinsed with an additional homogenization buffer to achieve a final volume of 6 mL. The filtered solution was centrifuged at 900 rcf for 10 min, after which the supernatant was discarded, leaving 50 μL of buffer above the pellet. The pellet was resuspended in 200 μL homogenization buffer, yielding a final volume of 250 μL. Next, the suspension was mixed 1:1 with 50% iodixanol (OptiPrep Density Gradient Medium, D1556, Sigma) prepared in 60 mM Tris (pH 8), 250 mM sucrose, 150 mM KCl, and 30 mM MgCl2. This mixture was carefully layered over 500 μL of 29% iodixanol in a 1.5 mL tube and centrifuged at 13,500 rcf for 20 min. Following centrifugation, the supernatant was gently removed to avoid disrupting the pellet. The pellet was resuspended in 50 μL of chilled, nuclease-free blocking buffer containing 1 × PBS, 1% BSA, and 0.2 U/μL RNasin Plus. This suspension was transferred to a fresh tube, brought to a total volume of 500 μL with blocking buffer, and prepared for fluorescent activated cell sorting (FACS). For neuronal enrichment, 1 μL of NeuN antibody (1:500, Millimark mouse anti-NeuN PE conjugated, FCMAB317PE, Merck) was added to the samples, which were incubated on ice in the dark for 30 min. After centrifugation at 400 rcf for 5 min, the supernatant was discarded, leaving approximately 50 μL of buffer above the pellet. The pellet was resuspended in 500 μL of blocking buffer, passed through a 20 μm filter into FACS tubes, and stained with 1 μL of DAPI (0.1 mg/mL, D3571, Invitrogen) prior to sorting.
Fluorescent-activated nuclei sortingThe nuclei suspension was maintained in darkness throughout the sorting process, which was developed using a flow cytometer (either BD FACSAria Fusion or BD FACSAria III) at 4 ℃. Gating was performed based on DAPI and phycoerythrin signals, separating the nuclei into two distinct populations: NeuN+ and NeuN−. Each sorted population was collected into separate tubes containing 50 μL of blocking buffer, with sorting continued until approximately 200,000 nuclei per population were obtained. Following sorting, the nuclei populations were centrifuged at 400 rcf for 4 min. The supernatant was carefully removed, leaving approximately 30 μL of buffer to resuspend the pellet. The samples were kept on ice to preserve their integrity for subsequent analysis.
Library preparationLibrary preparation from the sorted nuclei suspensions was carried out using the Chromium Next GEM Single Cell 3’ Reagent Kit v3.1 (PN-1000268, 10 × Genomics). Nuclei populations were manually counted, and their concentrations were adjusted to a range of 200–1700 nuclei/μL. Following the manufacturer’s protocol (CG000204 Rev D, 10× Genomics), reverse transcription (RT) mix was added to the nuclei suspensions. For library preparation, samples were either loaded into separate lanes on the Chromium Next GEM Chip G (PN-1000120, 10× Genomics) for each population (targeting a recovery of 5000 nuclei per population) or mixed prior to loading, with proportions of 70% NeuN + and 30% NeuN − nuclei (targeting a recovery of 5000 or 7000 nuclei). Subsequent cDNA synthesis and library preparation were conducted in accordance with the manufacturer’s instructions, utilizing the Single Index Kit T Set A (PN-1000213, 10× Genomics). Quality control and quantification steps within the protocol were performed using the Agilent High Sensitivity DNA Kit (5067–4626, Agilent Technologies) and the KAPA Library Quantification Kit (2700098952, Roche) to ensure the accuracy and reliability of the prepared libraries.
Illumina sequencingPooled libraries were prepared by combining up to 19 samples for a target nucleus recovery of 5000 or up to 16 samples for a target recovery of 7000 nuclei. Sequencing was conducted on a NovaSeq S6000 platform using an S4–200 (v1.5) flow cell with eight lanes, employing a 28–8–0–91 read configuration. The sequencing was carried out at the National Genomics Infrastructure in Stockholm, Sweden.
sn-RNA-seq data analysisPre-processingRaw single-nuclei RNA sequencing (sn-RNA-seq) data were processed into count matrices using CellRanger (v3.0.0, 10 × Genomics). The sequencing reads were aligned to the hg38 genome (GRCh38.p5; NCBI:GCA_000001405.20), incorporating both intronic and exonic sequences during the alignment process.
Quality controlTo identify potential doublets, we used Scrublet [84] for each sample individually, conducting 100 iterations with automated threshold detection, default parameters, and a fixed random seed. Nuclei identified as doublets in more than 10% of the Scrublet runs were excluded. Quality control based on the distribution of unique molecular identifiers (UMIs) and unique genes detected per nucleus was then performed. Nuclei with fewer than 500 UMIs or 1200 genes were removed, as were those with more than 250,000 UMIs, over 15,000 genes, or exceeding 10% mitochondrial content. For the remaining nuclei, we modeled the logarithmic relationship between the number of unique genes and UMIs as a second-degree polynomial function. Nuclei exhibiting significant deviations from the polynomial fit—defined as a difference greater than 2000 between log-transformed gene counts and the predicted value based on UMI counts—were classified as outliers and excluded. In addition, nuclei expressing high levels of marker genes corresponding to multiple cell types were removed. To assess this, a cell type score was calculated for each subtype (Oligodendrocytes, Microglia, OPCs, Neurons, Astrocytes, Vascular cells) for each nucleus, based on the mean expression of canonical marker genes. The score distributions were evaluated across the dataset, revealing bimodal patterns in all cases. These distributions were modeled as Gaussian mixtures, and a threshold was set at the mean of the lower Gaussian distribution plus four standard deviations. Nuclei exceeding the threshold for multiple cell types were classified as doublets and excluded. To further reduce contamination from neighboring regions, such as the claustrum or amygdala, nuclei expressing regional marker genes (NEUROD2, TMEM155, CARTPT, SLC17A7) were removed, with markers derived from the Allen Brain Atlas [32]. The number of nuclei excluded at each stage of this filtering process is summarized in Supplementary Fig. 1.
Oligodendrocytes detectionCount matrices were analyzed using Scanpy [82] to facilitate clustering and cell type annotation, with a focus on identifying oligodendroglia. Principal component analysis (PCA) was performed, and a neighborhood graph was computed using the first 30 principal components (PCs). Clustering was conducted using the Louvain algorithm at a resolution of 0.2. The resulting clusters were annotated as either glial cells or neurons based on the expression profiles of canonical marker genes:
Astrocytes —AQP4, ADGRV1
Microglia—CSF1R, FYB1
Oligodendrocytes—MBP, MOG, MAG
OPCs—PTPRZ1, PDGFRA, VCAN
Vascular cells—EBF1, ABCB1, ABCA9
Neurons—MEG3
Oligodendrocytes and OPCs were selected, and genes were filtered based on their expression per cell. Genes detected in less than three cells were removed (1389 genes).
Oligodendroglia lineage classificationOligodendroglia nuclei were projected onto the first 15 PCs calculated on their 10% of most variable genes and re-clustered using the Louvain algorithm. Harmony [43] algorithm was used to remove the batch effect and integrate the data. The function scanpy.tl.rank_genes_groups from Scanpy [82] was used to perform a differential expression analysis between the clusters through a Wilcoxon rank-sum test. Marker genes were selected manually from the top ranked genes (log fold change > 0.5 and p value < 0.05) to characterize and name each of the oligodendroglia clusters as a different oligodendroglia subclass. Markers for each subpopulation are represented in Supplementary Table 2.
Hierarchical analysisHierarchical clustering of the identified subpopulations was performed using the scanpy.tl.dendrogram function from the Scanpy [82] package. The clustering was based on the Pearson correlation method to measure similarity between subpopulations, and the complete linkage method was applied to define the cluster structure. This approach allowed for the visualization of relationships between subpopulations based on their transcriptomic profiles.
Compositional analysisThe differences in cellular proportion composition between conditions and brain regions were analyzed using the single-cell compositional data analysis (scCODA), a Bayesian model specifically designed to analyze changes in compositional data, particularly from single-nuclei RNA sequencing experiments. This method accounts for the inherent dependencies in compositional data, providing more accurate insights compared to both compositional and non-compositional alternatives. scCODA has been demonstrated to outperform other approaches in this domain, as described by Büttner et al. [14].
Module analysisTo identify specific gene modules associated with oligodendroglia subpopulations, particularly those linked to OPALIN expression, we applied Hotspot [20] to our subsetted dataset. Hotspot detects gene modules by leveraging co-expression patterns and analyzing single-nuclei transcriptomic data, enabling the identification of functionally related gene clusters. We used default parameters but restricted our analysis to the top 10,000 highly variable genes to focus on the most relevant features of the dataset.
Gene set enrichment analysisGene set enrichment analysis (GSEA) was performed using the Python package GSEApy (version 1.0.4). Pathways were considered significantly enriched if they exhibited a Normalized Enrichment Score (NES) greater than 0.5 and a false discovery rate (FDR) q-value below 0.05, to account for multiple hypothesis testing. The analysis was conducted using Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Reactome databases as reference sets for pathway annotation and functional enrichment.
GSEA superpathwaysGSEA results from all comparisons were processed in Python to generate superpathways and summarize enrichment patterns. Redundant pathways across contrasts were merged, with summary metrics including mean NES, average FDR, and gene coverage calculated for each. To evaluate pathway similarity, we adapted the code from https://github.com/ayushnoori/brainstorm/blob/master/R/pathway-clustering-functions.R in Python, calculating Jaccard indices based on gene set overlaps, and hierarchical clustering (average linkage, threshold = 0.8) was applied to group similar pathways. Each cluster (except for the top 10 with the highest and lowest NES, which were annotated manually) was annotated with mean values for statistics columns and gene and pathways content. To generate informative labels, we applied KeyBERT (initialized with allenai-specter) for unsupervised keyword extraction. Pathway names were used as input, and the top two ranked n-gram phrases (1–3 words) were selected as superpathway descriptors.
Correlation analysis (PDAO-1/PDAO-2/MOLA ratio vs MBP)To estimate correlations between subpopulations altered in PD (PDAO-1/PDAO-2/MOLA) and MBP values, we subsetted the data for samples showing values for all the variables (number of cells related to each subpopulation and MBP intensity). To be more precise, we calculated the percentage of cells in each sample related to PDAO-1/PDAO-2/MOL-A subpopulations and we only retained samples with more than five cells associated with a subpopulation. Finally, we used spearman correlation test with the following rationale: (PDAO-1 or PDAO-2 percentages per sample)/MOL-A and MBP intensity.
Trajectory analysisTrajectory analysis was conducted using two complementary methods. First, StaVia [68] was applied to integrate spatial and temporal data for mapping oligodendroglia differentiation. This method utilizes higher order random walks to trace lineage pathways and identify intermediate states. We executed the function run_VIA with default settings, modifying only the knn parameter to 15 for consistency across analyses. The second method, Slingshot [69], aligns single-cell data by identifying developmental lineages and connecting clusters to predict differentiation trajectories, effectively modeling the continuous maturation of oligodendroglia. Slingshot was also run with default parameters.
Factor analysisTo investigate the genes associated with the differentiation process, we employed factor analysis using the scikit-learn Python package. Factor analysis is a statistical method that models observed variables as linear combinations of potential underlying latent variables, known as factors. This technique aims to identify latent structures that explain the observed variability in the dataset while minimizing noise.
In this study, factor analysis was applied to pseudotime-marked gene expression data to uncover the primary latent factors driving gene expression changes during differentiation. The input data were normalized and scaled prior to analysis to ensure consistency and comparability across genes. By leveraging factor analysis, we identified key gene sets that contribute to distinct phases of the differentiation trajectory.
Communication analysisWith the aim of understanding the kind of communications and the quantity of them in oligodendroglia subpopulations, we used CellChat [37]. This method identifies ligand–receptor interactions across different subpopulations, constructing communication networks that reveal key signaling pathways involved in oligodendrocytes development. This analysis was developed in R, and we subsetted some populations, limiting the number of cells to 10,000 (comprising 5000 Control and 5000 PD randomly selected) in the case the number of cells of the subpopulation exceeded this number.
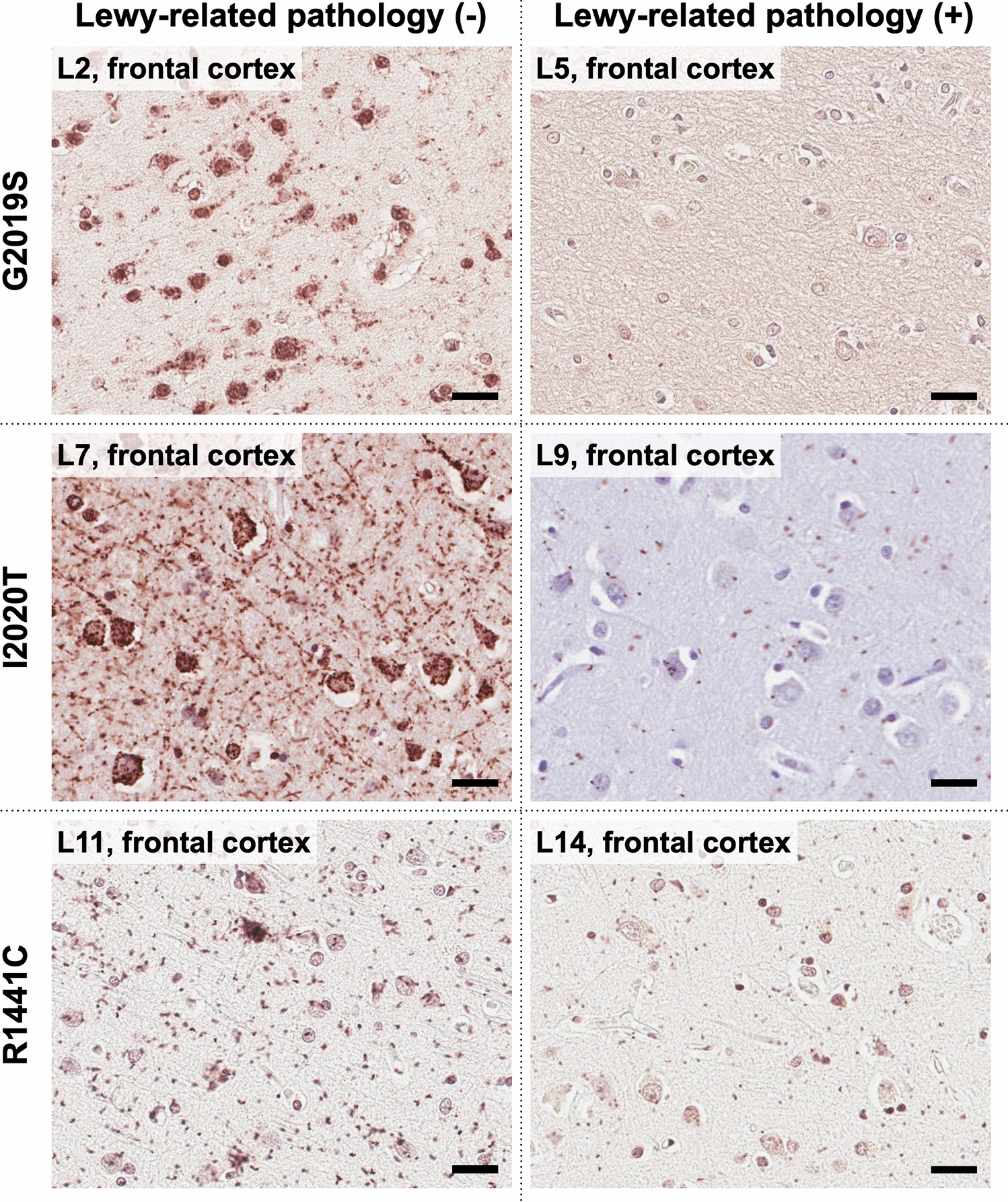
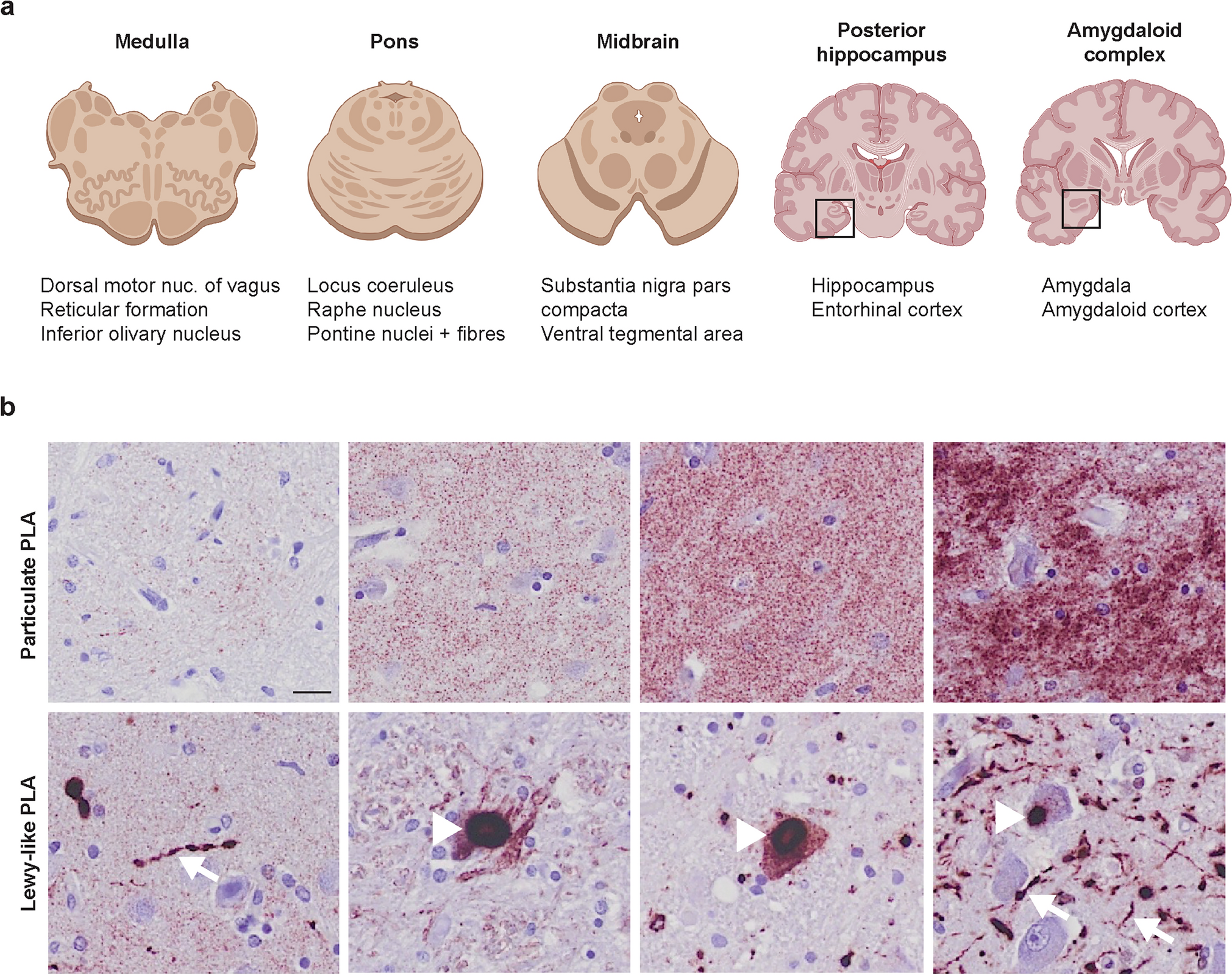
Immunohistochemistry with peroxidase-DAB methodEight-µm-thick formalin-fixed paraffin-embedded sections from the striatum (contralateral to samples used for sn-RNA-seq and spatial transcriptomics) at the level of the nucleus accumbens were immunostained in a Leica BOND RX fully automated research stainer using the BOND kit (Leica Biosystems, DS9800). Primary antibodies and concentrations were as follows: mouse anti-α-syn clone LB509 (Thermo Fisher Scientific Cat# 18–0215, RRID:AB_2925241, 1:200), mouse anti-myelin basic protein (MBP) amino acids 129–138 clone 1 (Millipore Cat# MAB382, RRID:AB_94971, 1:250), and rabbit anti-tyrosine hydroxylase (TH) polyclonal (Cell Signaling Technology Cat# 2792, RRID:AB_2303165, 1:100). The immunohistochemistry protocol included bake and dewax steps, a 20-min-long heat-induced epitope retrieval (with ER2 solution for α-syn and with ER1 solution for MBP and TH), and hematoxylin counterstaining. Sections were coverslipped with Permount mounting media and whole-slide images were obtained under the 40 × objective (numerical aperture 0.95, resolution 0.17 µm/pixel) of an Olympus VS120 virtual slide scanner (Olympus, Tokyo, Japan).
Immunohistochemistry images analysis and quantificationAll slide images were processed and analyzed using QuPath (version 0.4.3). Quantitative analysis focused on detecting intensity features related to MBP, as well as identifying the density of TH-positive projections from the SNpc to the dorsal striatum and α-synuclein aggregations (Lewy bodies/neurites, LBs/LNs). For each image, random regions of interest (ROIs) were selected within the putamen and caudate nucleus. TH-positive projections and α-synuclein aggregation detection was performed independently using QuPath’s pixel classifier tool. Five representative samples were chosen, and within each sample, the classifier was trained using 25 TH-positive and α-syn-positive annotations and 25 TH-negative or α-syn-negative annotations, resulting in a total of 125 TH-positive and 125 TH-negative annotations, and 125 α-syn-positive and 125 α-syn-negative annotations for classification.
The data obtained from MBP intensity measurements, TH projections quantification, and α-syn aggregate quantifications were further analyzed using the SciPy [78] Python library. A two-sample t test was applied to determine the T and p values for each brain region and condition. For MBP analysis, the mean of diaminobenzidine (DAB) intensity per pixel within the selected ROIs was used as the metric for quantification. TH differences were assessed by comparing the percentage of positive TH annotations, calculated as the area of TH-positive measurements, divided by the total area analyzed (µm2). Similarly, α-syn differences were quantified based on the proportion of α-syn-positive regions relative to the total area (mm2) examined in Pu or CN, respectively. In addition, Spearman’s rank correlation analysis was performed to explore relationships between various feature pairs across different regions and experimental conditions.
Spatial transcriptomics (Xenium)Spatial transcriptomics platformSpatial transcriptomic analyses were conducted using the Xenium high-plex in situ platform (10 × Genomics), which facilitates subcellular-resolution characterization of RNA within tissue sections, similarly as previously done in Garma et al. [28].
Gene panel designThe Xenium technology employs oligonucleotide probes designed to quantify the expression of genes in a predefined panel. For this study, a gene panel comprising 266 genes from the Xenium Human Brain Gene Expression Panel was utilized. An additional 100 genes were selected based on our sn-RNA-seq dataset, generating a custom gene panel (Xenium Custom Gene Expression Panel 51–100 (Z3DREH), PN-1000561, 10× Genomics).
Experimental workflowTissue blocks from human subjects (N = 4; including Pu and CN) were retrieved from − 80 ℃ storage and transferred on dry ice to a cryostat (CryoStar NX70, Thermo Scientific). Samples were mounted on the specimen holder using Tissue Tek O.C.T. Compound (4583, Sakura) and allowed to acclimate to − 20 ℃ within the cryostat chamber for 5 min. Tissue sections of 10 μm thickness were obtained and placed directly within the imaging region of precooled Xenium slides (12 × 24 mm, PN-3000941, 10× Genomics). Adherence of the sections to the slides was facilitated by briefly warming the reverse side of the slides with gentle pressure, followed by immediate refreezing on the cryobar. Tissue-mounted slides were retained in the cryostat during the sectioning process and subsequently stored at − 80 ℃. Downstream processing, including probe hybridization, ligation, and rolling circle amplification, was conducted at the in situ Sequencing Infrastructure Unit (Science for Life Laboratory, Stockholm), following the manufacturer’s protocol (CG000582 Rev E, 10× Genomics). Background fluorescence was minimized via chemical quenching. Tissue sections were imaged using the Xenium Analyser instrument (10× Genomics), which also facilitated signal decoding and data acquisition.
Oligodendroglia identificationXenium experiments provide spatial positions of all decoded reads. Therefore, the first step in processing these datasets is to segment individual cells and identify its composition. In this study, cells from Xenium experiments were defined based on default nuclear Xenium segmentation. This conservative approach minimizes the possibilities of missegmentation, as illustrated by Marco Salas et al. [45]. Segmented cells were then preprocessed using Scanpy to identify oligodendroglia. Essentially, pre-processing of the spatial dataset involved filtering out low-quality cells. Cells with fewer than 40 total counts or fewer than 10 detected genes were excluded. The data were then normalized, log-transformed, and subjected to neighbor graph construction using Scanpy’s pre-processing pipeline. A UMAP embedding was generated to visualize clusters, and Leiden clustering was applied with a resolution of 0.8 to identify distinct cell populations.
Oligodendroglia clusters (clusters 0, 3, and 9) were identified based on the expression of known oligodendroglia marker gene expression (MBP, OLIG1, OLIG2, PDGFRA, among others). The selected clusters were subsetted into a new AnnData object for further analysis. Marker gene expression was visualized spatially using matplotlib and scanpy to confirm the presence and distribution of oligodendrocytes.
Oligodendroglia cell type assignmentCell type deconvolution was performed using the cell2location [42] package, which infers the spatial distribution of cell types by integrating sc-RNA-seq data as a reference. First, the single-cell reference dataset, generated in this study, was preprocessed to define a knowledge base of oligodendroglia cell type-specific gene expression profiles. Genes were filtered to retain those expressed in at least five cells, representing at least 3% of the total dataset, and exceeding a mean expression cutoff of 1.12. The spatial dataset, including only predefined oligodendroglia cells, was filtered to retain genes that overlapped with the single-cell reference. Cell2location’s setup function was used to prepare the spatial AnnData object for model training, specifying the sample batch as a key parameter. A regression model was trained on the reference data to estimate cell-type-specific mean expression levels. The spatial data were then analyzed using the cell2location model, which was trained for 3000 epochs with a detection alpha parameter of 200 and batch size of 10,000. Posterior distributions of cell abundances were exported to generate quantitative summaries of cell type probabilities for each spatial location. The q50 quantile (median) cell type abundances were visualized and further analyzed to identify the dominant cell types in each spatial coordinate. For oligodendrocytes, additional analysis of spatial localization and confidence intervals was performed using the posterior distributions.
Neighborhood enrichment analysisTo assess the neighborhood enrichment of oligodendrocytes, spatial interaction networks were constructed for each sample in the dataset. The AnnData object containing oligodendroglia profiled with Xenium was first subsetted by sample, and spatial neighbors were computed using Squidpy’s [53] spatial_neighbors function with a radius of 124 um. This process was repeated for all samples, and the resulting AnnData objects were concatenated into a unified dataset.
Neighborhood enrichment was calculated using Squidpy’s nhood_enrichment function, with the cell type annotation, defined using cell2location, as the cluster key. This analysis computes z-scores representing the degree of enrichment or depletion of specific cell types in the neighborhoods of other cell types. Visualization of neighborhood enrichment was performed using a heatmap with a diverging colormap (coolwarm), highlighting significant positive and negative interactions. Parameters such as color scale limits (vmax = 200, vmiN = -200) and figure size were adjusted to optimize clarity and interpretability. Overall, this method provides insights into the spatial interactions and organization of oligodendrocytes in their local tissue environment, enabling a deeper understanding of their roles in tissue architecture and function.
Module identification using hotspotWe used Hotspot [20] to identify gene expression modules in oligodendroglia profiles derived from Xenium spatial transcriptomics data. Hotspot identifies informative genes and gene modules based on how gene variation aligns with cell similarity metrics. Informative genes show expression patterns concordant with local cell similarity. In summary, we initialized Hotspot with the following parameters:
Layer key: ‘raw’ for raw expression values.
Model: Negative binomial (danb).
Latent obsm key: ‘X_pca’.
UMI counts key: Total UMI counts per cell.
A k-nearest neighbors (kNN) graph (20 neighbors, unweighted) was created to compute gene autocorrelations. Genes with FDR < 0.05 were selected as significant. Local correlations for these genes were computed using parallel processing. Gene modules were defined using a minimum threshold of five genes, with core genes meeting an FDR threshold of 0.05. Module scores for each cell were calculated and stored. Differences in module scores between PD and control groups were analyzed using the Wilcoxon rank-sum test. Violin plots visualized score distributions, with test statistics and p values annotated.
Niche identification using NicheCompassNicheCompass [8] was used to identify cellular domains by integrating spatial transcriptomics data across all samples. A latent graph embedding was computed using a graph convolutional network (GCN) model with categorical covariates such as replicates. NicheCompass was run for 400 epochs with regularization to optimize ligand-receptor-based niche identification. Clustering was performed on the latent embeddings using the Leiden algorithm with a resolution of 0.2. After clustering, niches with fewer than 200 cells were filtered out, leaving three robust niches, that could be annotated as white matter (WM), gray matter (GM), and vascular niches based on their expression and cellular composition. These niches were characterized by their communication program activity, spatial localization, and enriched gene targets.
Statistical analysisStatistical analyses were conducted using Python-based computational tools. Differential expression analysis was performed using the Wilcoxon rank-sum test, with marker genes selected based on a log fold change > 0.5 and p value < 0.05. Hierarchical clustering of subpopulations was conducted to assess similarity based on Pearson correlation. Cellular compositional differences between conditions and brain regions were analyzed using a Bayesian model designed for sn-RNA-seq data. GSEA statistical significance was assessed using permutation-based methods (a Kolmogorov–Smirnov-like statistic), which compares the observed enrichment of genes in a gene set against a random distribution. Spearman’s rank correlation was used to examine relationships between specific features, such as the percentages of PDAO-1 or PDAO-2 and MOL-A/MBP intensity. Two-sample t tests were used to compare MBP intensity, TH-positive dots, and α-syn-positive areas across conditions and brain regions, generating T and p values.
Sample sizes were determined based on prior studies and experimental feasibility. Detailed statistical results and methodologies are presented in the figures and legends.
Comments (0)