
Remember me


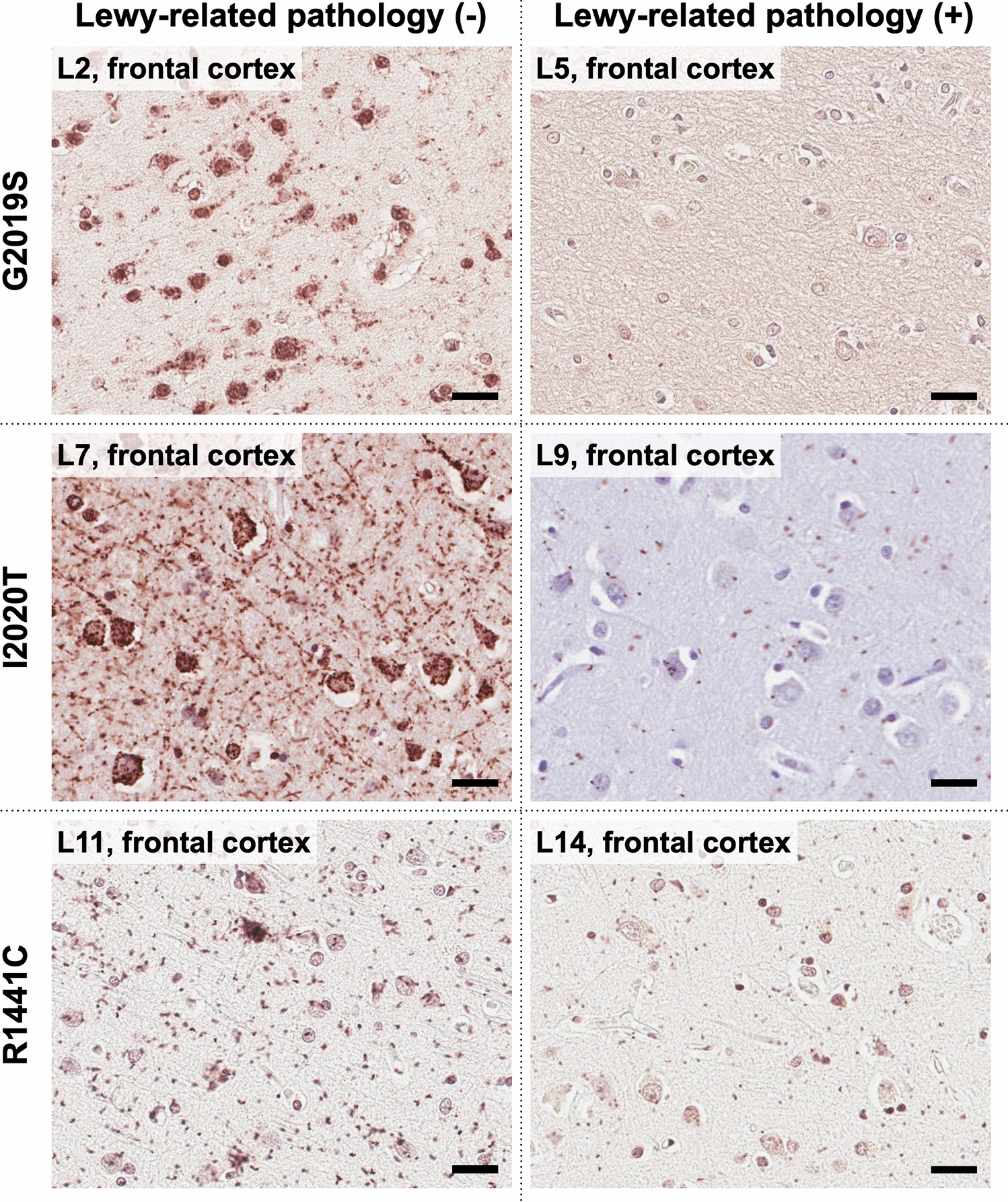
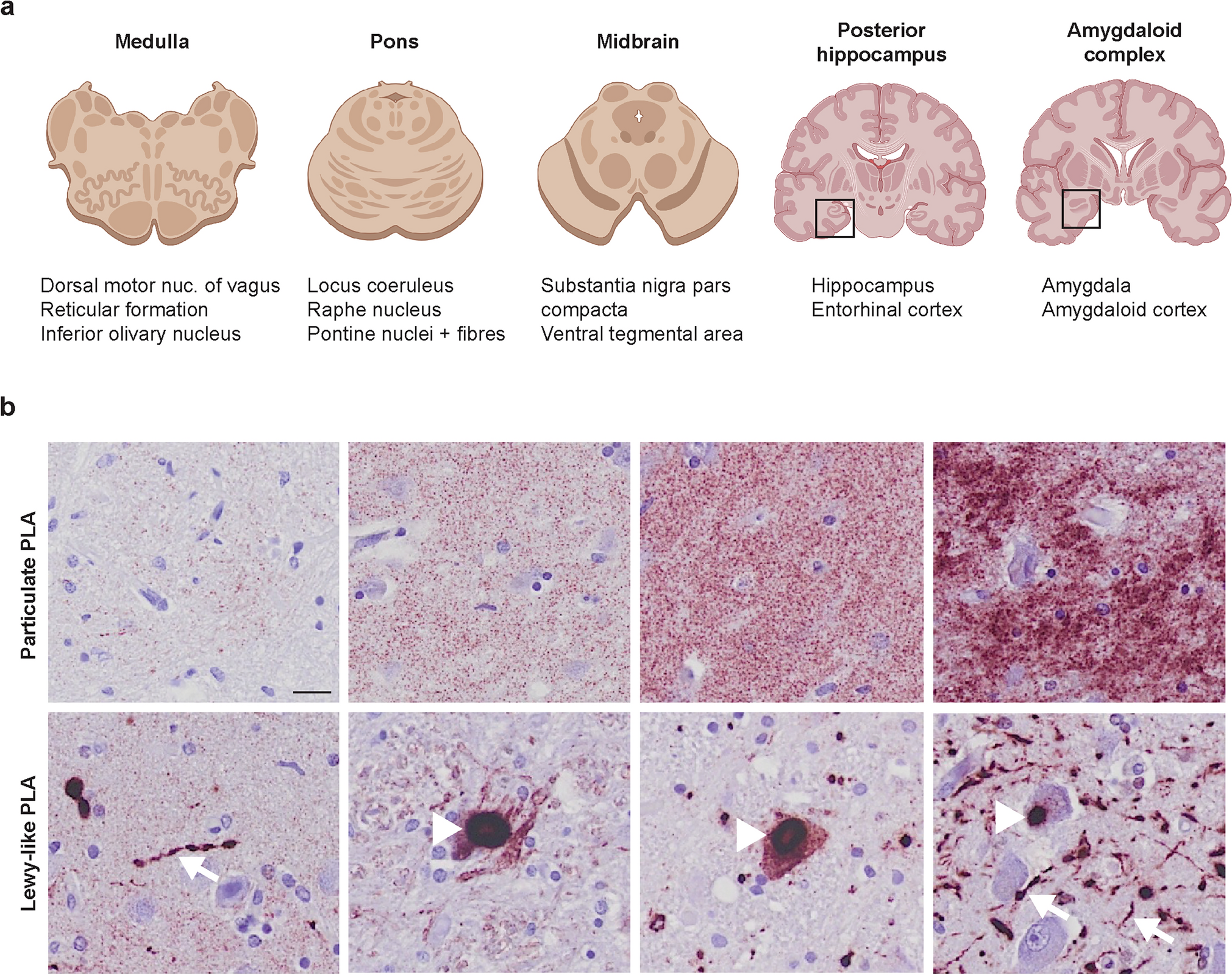



Several genes that potentially increase risk for PSP have been detected from association studies (Table 1). Genes found by associations are listed and their function is discussed if their association reached genome-wide significance.
Table 1 Genes conveying risk for PSP(P < 5E-8). For any gene that conveys risk we first describe the gene and its function and subsequently report and appraise observations of its risk for PSP.
Before going into the detailed findings of gene associations we want to point out a few caveats of gene assignments by association with SNPs. For example SNPs in high linkage disequilibrium regions may tag multiple genes and the assignment of the “correct” gene might be difficult [28, 97]. Furthermore SNPs are usually located in non-coding regions and might affect distant regulatory regions rather than the nearest gene [28, 97]. This said we describe the findings.
MAPT encodes microtubule-associated protein tau and is highly expressed in neurons and to a lesser extent in oligodendrocytes. The MAPT gene is located in 17q21.31 (nucleotide positions: 45,894,527–46,028,334) and is composed of 16 exons. Exons 2,3,10 are alternatively spliced leading to six isoforms of tau [7]. Exons 9–12 contain four repeat sequences that code for the microtubule binding domains of tau. These domains are composed of imperfect repeats of 31 or 32 amino acids (four highly conserved blocks of 18 amino acids, separated by either 13 or 14 different amino acid residues) in the carboxyl terminus of the protein [34, 95]. Exclusion of exon 10 results in tau proteins containing 3 imperfect repeats (3R, −10) and inclusion of exon 10 gives rise to tau proteins containing 4 repeats (4R, + 10). There are two major haplotypes, H1 and H2, spanning approximately 1.8 Mb which contain the entire MAPT gene. H2 is defined by an inversion of approx. 900 kb relative to H1, thus avoiding recombination between the two haplotypes [14]. H1 is much more common than H2 and H2 expression is lower than expression of H1 [78]. H1 and H2 include several specific single nucleotide polymorphisms (SNPs) [72]. Based on H1-specific SNPs at least 23 H1 sub-haplotypes have been described [38]. Tau binds to and stabilizes microtubules in neurons. By interacting with ribosomes tau regulates mRNA translation and transports proteins in axons. It influences the function and stability of synapses [35] and is involved in neuronal differentiation and function [10]. Tau is mainly synthesized by neurons but also by oligodendrocytes and astrocytes.
The MAPT region (Fig. 1) extends beyond the gene proper and contains two genes (FMNL1 and ARHGAP27) that function as cis-regulatory elements (CREs) of MAPT. These CREs operate as enhancers and interact with MAPT promoters in neurons. There are two regions at the FMNL1 (Formin Like 1 with at least 3 isoforms; https://www.uniprot.org/uniprotkb/O95466/entry) locus that appear to regulate MAPT expression. These two regions interact with the MAPT promoter in both excitatory and inhibitory neurons [78]. Interestingly, expression of the FMNL1 gene is very low in the brain. FMNL1 obviously has two functions, one as an enhancer of MAPT in cells of the brain and another one as a protein coding gene in other tissues. Furthermore, there are two additional regions containing ARHGAP27 (encoding Rho GTPas activating protein 27 with at least 4 isoforms; https://www.uniprot.org/uniprotkb/Q6ZUM4/entry), 464,677 bp and 461,949 bp upstream of MAPT that have enhancer function [78]. In addition, there appears to be an intronic CRE within MAPT. Another gene in the region, MAPT-AS1 (MAPT antisense RNA 1, an RNA gene belonging to the lncRNA class) might operate as an epigenetic regulator of MAPT expression [19, 78].
Fig. 1
MAPT region in 17q21.31. Genes discussed in this article are indicated by arrows and their DNA coordinates are given. (adapted from the UCSC Genome Browser at http://genome.ucsc.edu)
STH and KANSL1 that lie within or close to MAPT (figure) are co-expressed with 4RMAPT at least in brains of Parkinson disease cases [93] suggesting interacting enhancer functions.
MAPT is associated with PSP risk as demonstrated by numerous findings:
1)About 15 different point mutations of MAPT have been detected in families with autosomal dominant inheritance of cases presenting as PSP. One of these mutations (p. L284R) is located in exon 10 [89], another one (p.E342 K) in exon11 [56] of the gene. Additional point mutations have been found in atypical PSP and FTDP-17 mainly in exon 10 of MAPT [8, 46]. The cases with PSP-like presentations and point mutations appear to differ from classical PSP.
2)While 3R and 4R are present in normal brains in equal amounts [29], 4R tau is preferentially found in PSP [30].
3)Of the two major haplotypes of MAPT, H1 is associated with increased risk for PSP and H2 is protective [31, 32]. The frequency of H1 is 95% in chromosomes of PSP patients as compared to 77.5% in control chromosomes [33, 34].
4)The H1 sub-haplotype H1c is overrepresented in PSP as compared to controls. H1c is defined by presence of an A nucleotide of the reference SNP rs242557 14. This SNP is located in the regulatory region of MAPT and allele A is thought to trigger higher expression of MAPT. This is likely to increase the amount of tau protein and thus the risk for PSP and other tauopathies such as corticobasal degeneration (CBS) [38]. Several additional sub-haplotypes of H1 (H1 d, H1 g, and H1o) are also significantly increased in PSP as compared to controls [38].
5)There are three large copy number variants (CNVs), designated α, β, and γ within H1c. While α, β do not affect the risk for PSP increased copies of γ enhance PSP risk [102].
6)Genetic association studies found highly significant association of polymorphic sequences in non-coding regions of MAPT with PSP, such as SNPs rs8070723 and rs242557 [43, 81]. A meta-analysis of two genome wide association studies [43, 81] yielded genome wide significance of P = 5.51E-144 for rs.8070723 and of P = 3.78E-85 for the association of PSP with rs242557. rs8070723 is located in an intron of MAPT [3] and rs242557 is located in a highly conserved repressor domain in the MAPT promoter region [6]. Tau levels are increased in all brain areas affected in PSP [44]. It accumulates in neurons, oligodendrocytes and astrocytes [29]. Up-regulation of MAPT contributes to the development of PSP by increasing the amount of tau and altering its posttranslational modifications (methylation, phosphorylation) and interfering with its normal functions [30]. For example elevation of tau might disturb its effect on stability of microtubules which would eventually result in cell death. In the brain the increase of tau might disturb the stability of synapses and might interfere with the function of neurons. Transcriptomic analyses showed that the increase in tau is mainly due to haplotype-dependent increased expression of 4RMAPT [76].
7)ARHGAP27 expression is dysregulated in cerebellum and temporal cortex in PSP. Since dysregulated co-expression with MAPT was described and MAPT transcription is increased at least in the frontal cortex of PSP, this might indicate that expression of ARHGAP27 is upregulated in given brain structures as well [76].
There are several genes in the MAPT H1/H2 inversion region such as KANSL1, PLEKHM1 (P = 1.0E-9) LRRC37A4 (P = 2.2E-22), ARL17A (P = 9.2E-22) that are — as is MAPT (P = 8.71E-28) — significantly associated with SNPs within this region, in particular rs.242557. It was discussed [43] that association with markers in this region reflects proximity to MAPT. This is indeed the case (Fig. 1). A recent study investigated whether these genes are regulated similar to MAPT. Analysis of eQTLs in the H1/H2 region demonstrated that some genes in close proximity to MAPT are regulated independent of MAPT [26, 76]. Two of these genes are KANSL1 and PLEKHM1.
KANSL1 (KIAA1267) is located in the MAPT region of chromosome 17q21.31 (Fig. 1) and encodes the nuclear protein KAT8 Regulatory NSL Complex Subunit 1 (NSL1). This protein is a component of a protein complex, MLL1 and NSL1 which are involved in histone acetylation and methylation [67]. These histone modifications are probably the underlying mechanisms for their roles in mitosis, cell proliferation, and enhancer regulation [84]. Specifically, NSL1 regulates the fusion of autophagosomes and lysosomes [57]. In the mouse, reduced activity of KANSL1 interferes with clearance of impaired mitochondria which results in an increase in reactive oxygen species and eventually in neuronal death [58]. In humans, expression of KANSL1 is highest in the cerebellum. Among single cells in the brain expression is highest in astrocytes and lowest in vascular cells (https://v18.proteinatlas.org/ENSG00000120071-KANSL1/tissue).
Findings in RNA libraries from cerebellum of PSP patients and controls suggest differential expression of KANSL1 and MAPT [26, 76]. These observations are consistent with those of a previous study [87] that also indicated independent expression of KANSL1 and MAPT. Therefore KANSL1 might convey risk for PSP independent of MAPT. KANSL1 is increased in oligodendrocytes of the temporal cortex and the cerebellum of PSP in a haplotype (H1)-dependent manner [26, 76]. Although clearance of impaired mitochondria is impeded owing to a decrease in KANSL1 activity [57], an accumulation of NSL1 might also impede autophagosome–lysosome fusion, such as the promotion of the degradation of cellular components. While KANSL1 deficiency affects synaptic function by disturbed autophagy, increased transcription of the gene might have a similar effect [58].
PLEKHM1 encodes Pleckstrin Homology and RUN Domain Containing M1 (PKHM1). It is located on human chromosome 17q21.31 in the MAPT region (Fig. 1). PLEKHM1 is involved in the fusion of endosomes and lysosomes and in late stages of endolysosomal maturation. It may also play a regulatory role in autophagic and endocytic trafficking. Mutations in this gene have been found in patients with osteopetrosis [99]. PLEKHM1 is expressed in most tissues. In the brain, highest expression is in the cerebral cortex and cerebellum, and medium expression in hippocampus and nucleus caudatus, and hypothalamus. Expression is highest in oligodendrocytes, followed by astrocytes, microglia, and neurons and is lowest for vascular cells (https://v18.proteinatlas.org/ENSG00000225190-PLEKHM1/tissue). PLEKHM1 interacts with Rab7 and suppresses endocytic transport [91]. In PSP PLEKHM1 expression is increased as compared to controls [76]. This might interfere with normal function of oligodendrocytes, e.g., by causing abnormal lysosomal metabolism and/or impaired autophagosome–lysosome fusion that eventually cause death of this cell type. Significantly, several additional genes such as MOBP, SLCO1 A1, NFASC, and CNTN2 (see below) are relevant for normal function of oligodendrocytes that is disturbed in PSP.
STX6 is located in the long arm of chromosome 1 (1q25.3) and codes for syntaxin-6. Syntaxin-6 is a SNARE (Soluble N-ethylmaleimide-sensitive factor attachment protein receptor) protein with various functions in cellular trafficking. It is involved in targeting of endosomal vesicles to the trans-Golgi network (TGN) and to a lesser extent from TGN to endosomal vesicles. It is also required for movement of vesicles from endosomes to the cell membrane [51, 106]. STX6 is expressed in most tissues, yet highest in hepatocarcinoma cell lines. In the brain it is expressed in oligodendrocytes and astrocytes, neurons, microglia, and vascular cells. Expression is highest in the cerebral cortex and somewhat less in cerebellum, hippocampus, and caudate nucleus (https://www.proteinatlas.org/ENSG00000135823-STX6/tissue).
STX6 appears to be involved in PSP. A significant association was found with an allele of SNP rs1411478 at position 180,993,146 (GRCh38 assembly) of 1q25.3 [43, 27, 81]. This SNP lies in intron 4 of STX6 [27]. Ferrari et al. [27] found significant association (P = 1.8E-9) of rs1411478 with PSP. In our cohorts [43] significance of association was P = 2.3E-10 with the same SNP. It may be that the disease-associated allele of STX6 influences movement of misfolded proteins from ER to lysosomes and thus may contribute to neurodegeneration. Based on specific down-regulation of STX6 in white matter of patients as compared to controls, Ferrari et al. [27] speculated that the associated allele of STX6 contributes to white matter pathology in PSP. This was shown on the basis that the associated risk allele rs1411478 is a strong expression quantitative trait locus. In a later study Farrell et al. (2024) [26] found that the lead SNP of their GWAS, i.e. rs1044595-C is associated with increased expression of STX6 in brain samples and in purified oligodendrocytes [26].
MOBP is located in the short arm of chromosome 3 (3p22.1) and codes for Myelin Associated Oligodendrocyte Basic Protein [65, 109]. MOBP is expressed in the white matter of the cerebral cortex, basal ganglia, hippocampal formation, midbrain, amygdala and hippocampus and — at very low levels of expression — in the cerebellum [109]. Expression is by far highest in oligodendrocytes and much lower in microglia and vascular cells, and hardly detectable in astrocytes. (https://www.proteinatlas.org/ENSG00000168314-MOBP/tissue#rna_expression). MOBP synthesized by oligodendrocytes plays an essential role in stabilizing the myelin sheath in the central nervous system.
MOBP is likely involved in PSP. SNP rs1768208 within an intron of MOBP was significantly (P = 1E-16) associated with PSP [26, 27, 43]. The risk allele results in higher expression of MOBP in patients as compared to controls [3]. The authors speculate that increased levels of MOBP result in neuropathology of oligodendroglia thus contributing to PSP risk. Performing eQTL analyses a more recent study [26] found decreased expression of MOBP in both frontal cortex and cerebellum. The different results concerning the regulation of MOBP expression are likely due to the two different methods applied. While upregulation was found in associations between PSP risk variants and temporal cortex levels of 20 genes within ± 100 kb [3], decreased expression was found by eQTL analysis [26].
EIF2AK3 is located on chromosome 2p11.2 and encodes Eukaryotic Translation Initiation Factor 2-α Kinase 3 (PERK). It is a metabolic stress-responsive kinase that phosphorylates eIF2α (α subunit of eukaryotic translation initiation factor 2). PERK halts protein synthesis when unfolded proteins accumulate in the endoplasmic reticulum (ER) (unfolded protein response, UPR), thus preventing ER stress [90, 111]. It is expressed in oligodendrocytes and to a lesser degree in astrocytes and neurons. Its highest expression is in vascular cells.
Comments (0)