
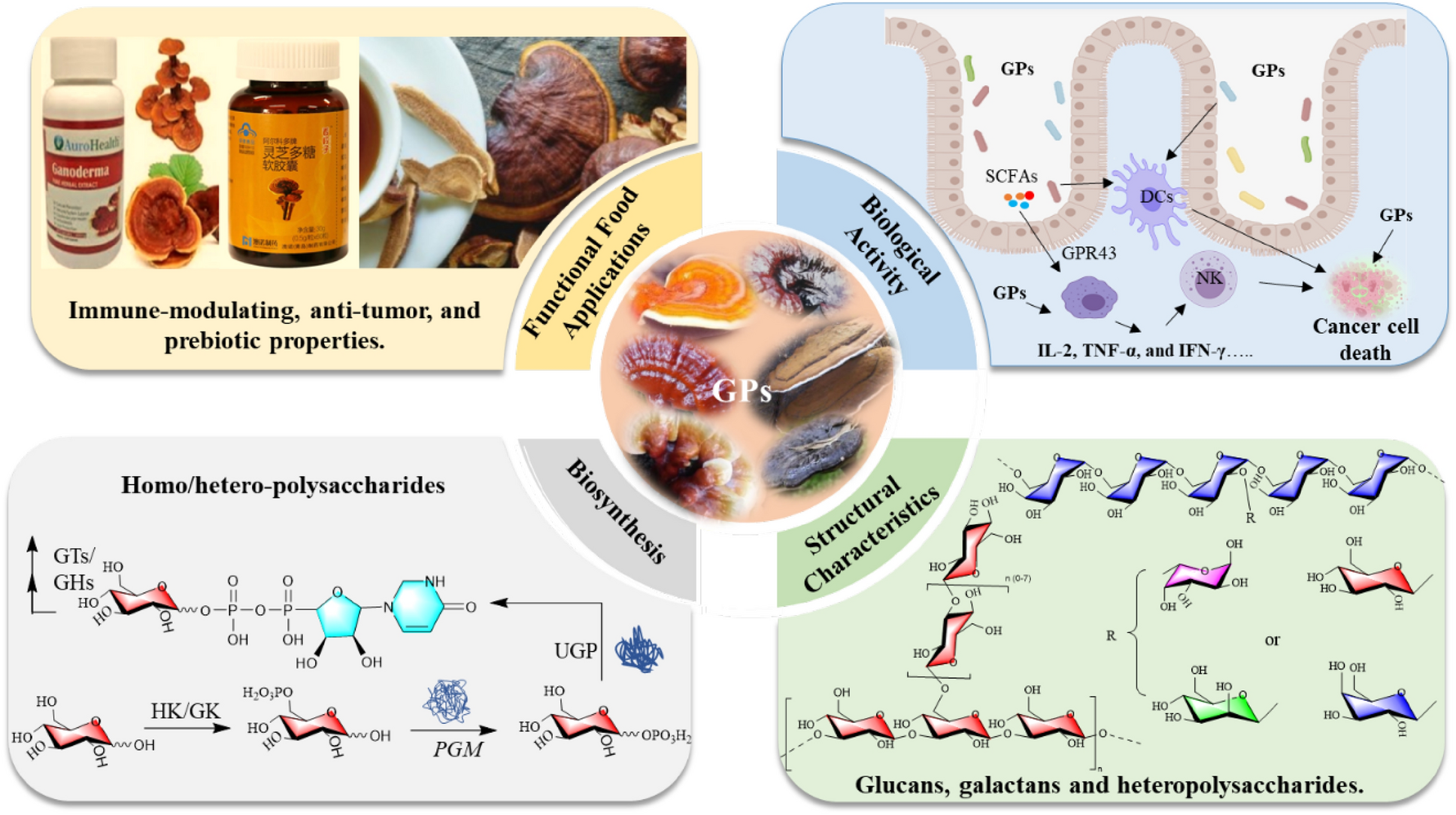
Remember me


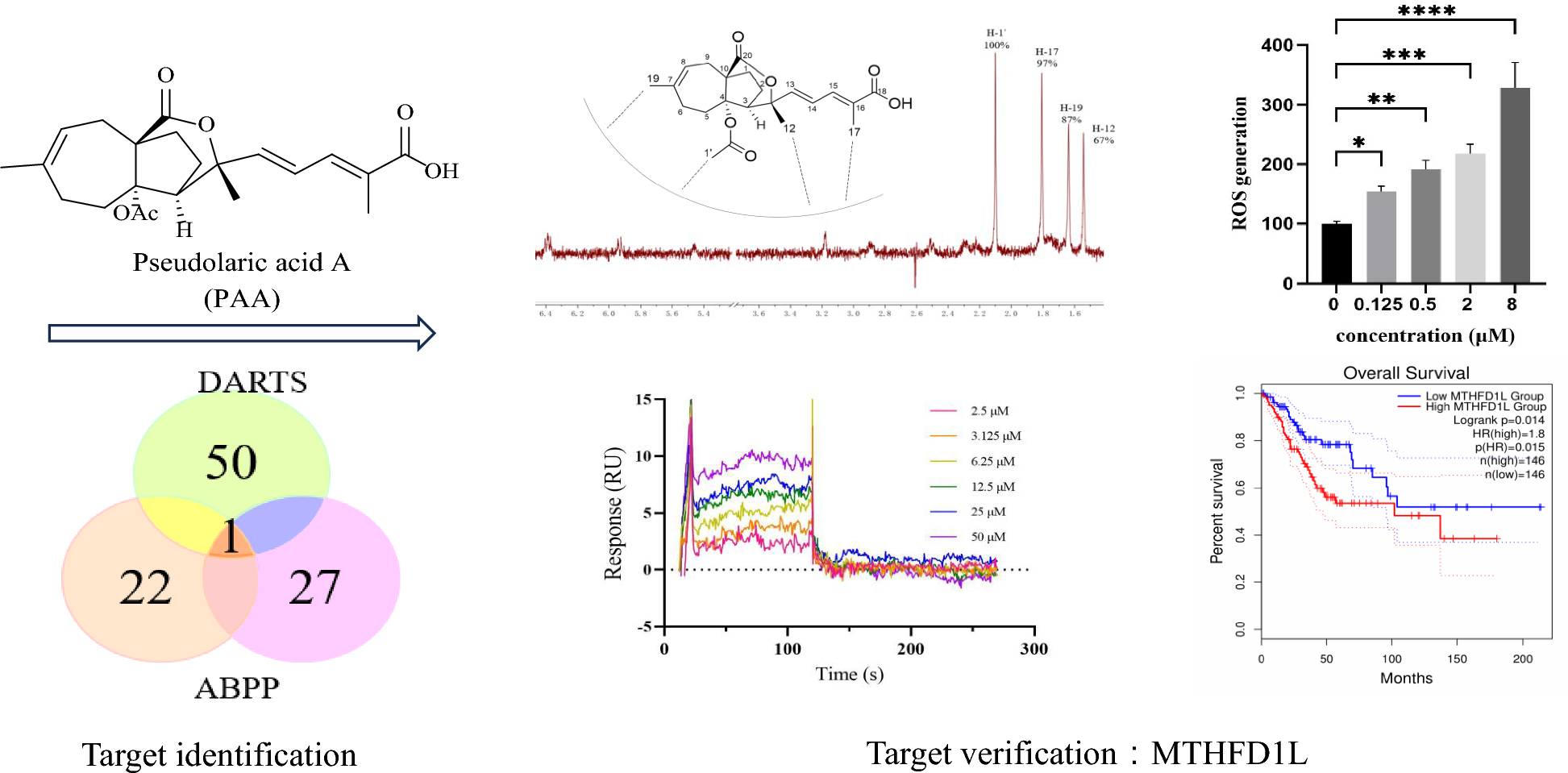

Considering that PAA exhibited significant antitumor activity in HeLa cell (cervical cancer cell line) (IC50 = 2.92 ± 0.36 µM), we conducted the label free target identification method based on shift in the thermal stability of protein, which is DARTS-centered technology to explore the protein targets of natural product PAA in HeLa cells. Different ratios of pronase to proteins were applied to optimize the hydrolysis conditions (Fig. 1B). When small compounds bind with proteins, this interaction stabilizes the target proteins, thus making them resistant to protease. The LC–MS/MS analysis of the digested peptide samples and Mascot database search could identify the potential binding proteins. The list of putative PAA interacting partners was refined by only including proteins protected by PAA. Results from two repeats of the DARTS experiments showed that there were 42 overlapping proteins, both of which were up-regulated in the PAA and pronase-treated group, as the result of PAA induced protein stabilization and reduction in proteolysis susceptibility. Part of the potential binding proteins was shown in Fig. 1C. The initial identification of protein targets using DARTS chemical proteomics approach exhibited 42 potential binding partners of PAA (Table S1).
Fig. 1
Potential binding proteins of PAA identified by DARTS. A Chemical structure of PAA. B Coomassie stained gel showing proteins with/without treatment of pronase. The HeLa cell lysate was treated with PAA and different concentrations of pronase. Different ratios of PAA to pronase were optimized, and 1:100 (pronase: protein sample) was chosen for the following procedures. C Part of the putative PAA interacting proteins, which has been identified with the following two criteria: fold change of different groups (Fold Change > 1.5) and p value < 0.05
2.2 Potential binding proteins of PAA identified by ABPP methodTo further refine the potential binding proteins of PAA, we further utilized the activity-based protein mass spectrometry (ABPP) strategy using the PAA-probe designed in previous study [10]. PAA probe exhibited similar biological activity as PAA (Fig. 2A). Dead probe was set as a negative control. The HeLa cell proteomes were divided into three groups to explore PAA interacting protein, including control group (10 μM dead probe), PAA-probe group (10 μM PAA-probe), Competition group (500 μM PAA first and followed by 10 μM PAA probe). When bands from SDS-PAGE displayed no fluorescent intensity in the control group, the highest fluorescent intensity in the probe-treated group, and weaker fluorescent intensity in the competition group, they are considered to exhibit apparent competitive effects. Bands ranging from 40–55 kDa, 70–80 kDa, and 90–130 kDa were cut and proceeded with quantification sequencing (Fig. 2B). The ABPP workflow was repeated twice to reproduce the results. 18 potential binding proteins were overlapped in the two repeated assays. The detailed information of the overlapped proteins was shown in Fig. 2C (Table S2).
Fig. 2
Potential binding proteins of PAA identified by ABPP. A Chemical structures of PAA-probe and dead-probe (control probe). B Vanne map shows the overlapping proteins through two repeats of ABPP assay (left) and competition assay of the PAA-probe and PAA alone, visualized by in-gel fluorescence scanning at 533 nm (right). C Collected protein and gene information of the overlapped binding partners of PAA from two repeats of the ABPP assay
2.3 Most potential binding target of PAA and its association with cervical & endocervical cancer through bioinformatic analysisAmong all potential binding targets of PAA detected by two different strategies, methylenetetrahydrofolate dehydrogenase 1 like (MTHFD1L) protein is the only overlapped target (Fig. 3A). We therefore propose MTHFD1L is the most potential binding target of PAA. MTHFD1L is the human mitochondrial C1-terrahydrofolate synthase, catalyzing the conversion of tetrahydrofolate (THF) to 10-formyltetrahydrofolate (10-CHO-THF) in mitochondria [16, 17]. It is the core enzyme in the one-carbon (1C) cycle metabolism, which is highly expressed in a variety of tumors and associated with tumor cell proliferation and invasion [18, 19].
Fig. 3
MTHFD1L is one of the most potential binding partners of PAA and highly involved with cancer through bioinformatic analysis. A Vanne group demonstrating the potential binding partners of PAA identified by DART and ABPP strategies. B The gene expression profile of MTHFD1L across all tumor lines and paired normal tissues. The height of bar stands for the median expression of certain tumor type or normal tissue. C The expression of MTHFD1L in CESC was analyzed through GEPIA2 database. The expressions of MTHFD1L in CESC patients (n = 306) and normal people (n = 13) in the TCGA database were analyzed. D The overall survival analysis of MTHFD1L in CESC cancer cell line through TCGA database showed the significant prognostic impact of MTHFD1L
To get a deeper understanding of gene MTHFD1L functions in tumor cells, we further mined the data through The Cancer Genome Atlas (TCGA) databases (https://www.cancer.gov/tcga) and Genotype-Tissue Expression (GTEx) database [20]. Clinical data from two databases demonstrated that MTHFD1L is differentially overexpressed in multiple cancer subtypes and paired normal tissues (Fig. 3B). Since PAA exhibited decent antitumor activity in HeLa cell (cervical cancer cell line), we paid extra attention to the expression profiles of MTHFD1L in cervical &endocervical cancer (CESC) line. Gene expression profile showed that MTHFD1L is highly overexpressed in CESC patients, compared to the expression in normal people (Fig. 3C). Furthermore, survival analysis allows the identification of correlation between gene expression and prognostic outcomes. We performed the survival analysis to evaluate clinical relevance of MTHFD1L gene through GEPIA2 [21]. Result was shown in Fig. 3D and we found that MTHFD1L exhibited significant association with unfavorable prognostic outcome in CESC cancer, but not in other cancers (Figure S2). Taken together, comprehensive bioinformatic analysis has aided the understanding of the important role of MTHFD1L gene in CESC cancer type and led to the identification of MTHFD1L as a potential therapeutic target and biomarker.
2.4 Verification of the direct interaction between MTHFD1L and PAAWe used nuclear magnetic resonance (NMR) saturation transfer difference (STD) and surface plasmon resonance (SPR) techniques to verify the direct interaction between MTHFD1L and PAA in the molecular level.
The ligand-observed NMR STD studies could determine the direct protein–ligand interaction and provide information on the putative orientation of ligand to target protein. Protons in the PAA closest to the protein upon binding show the strongest STD effect. The STD effect of PAA is shown in Fig. 4A. H-1’ gives the strongest enhancement and is considered to interact the most with the protein surface. This observation is consistent with the previous structure–activity relationship (SAR) analysis that H-1’ is essential for the activity of PAA. Both our previous data and reports from other groups have demonstrated that deacetyl derivatives of PAA exhibit no antitumor activity, indicating that the C4-acetoxy group is crucial for this activity [10, 22]. Notably, H-1’ is located within the C4-acetoxy group. Pseudolaric acid C (PAC), which lacks the -COOCH3 moiety and possesses a C4 hydroxyl group instead, is also inactive. H-17 and H-19 exhibited similar but slightly less STD effect, indicating the critical importance of these two groups in the MTHFD1L-PAA binding mode, in accordance with the SAR result. STD effects of H-12 and other protons suggest that these protons are not in direct contact with the protein. Taken together, NMR STD method demonstrated PAA directly interacts with target protein MTHFD1L in a defined orientation that the -OAc moiety and H-17 closely faces the protein surface.
Fig. 4
PAA directly interacts with MTHFD1L. A Chemical structure of PAA and its relative orientation with MTHFD1L revealed by NMR STD assay, with the 1H STD spectrum as red. B SPR assay showing the direct interaction between PAA and MTHFD1L. The steady state analysis method was used to fit the dissociation constant
We further verified and quantify the binding affinity of compound PAA to the target protein MTHFD1L through surface plasmon resonance (SPR) binding experiments (Fig. 4B). The equilibrium dissociation constant (Kd) is obtained by steady-state affinity analysis. Compound PAA and target protein MTHFD1L are fast binding and fast dissociation binding modes, and the equilibrium dissociation constant Kd is fitted, with Kd about 4.05 μM. The SPR results further confirmed the direct interaction between PAA and MTHFD1L.
2.5 Molecular docking analysis to generate a possible binding mode of the PAA to MTHFD1LTo gain the insight into the possible binding mechanism between PAA and MTHFD1L, molecular docking analysis with Autodock was performed. Due to the lack of published crystal structure, we adopted AlphaFold2 to predict the three-dimensional structure of MTHFD1L protein. The ligand with binding energies lower than − 7 kcal/mol was identifies as potential binding. The docking studies showed that PAA was well located within the predicted ATP—binding pocket of the MTHFD1L, with the binding energy of − 7.88 kcal/mol. This model with the lowest energy was chosen for subsequent protein–ligand interface analysis.
As shown in Fig. 5, H-1’ and H-19 are embedded in the center of the ATP binding pocket of MTHFD1L, suggesting its potential inhibition mechanism for MTHFD1L. C-19 forms hydrophobic interaction with residues M613, L616 and A617. The carbonyl group at C-4 (C-1’ group) forms hydrogen bond with the side chains of H678. C-17 methyl group forms interactions with the hydrophobic pocket formed by residues L485, P455 and V677 of MTHFD1L. Taken together, the hydrophobic interactions between protons in the PAA (C-1’, C-17 and C-19) and MTHFD1L strengthen the position of PAA in the ATP binding pocket of MTHFD1L. The groups of PAA highly involved in the interaction with MTHFD1L from molecular docking results are in line with the gathered experimental NMR STD data.
Fig. 5
Molecular docking analysis exhibits the binding interactions between PAA and MTHFD1L. Ligand PAA is denoted in green. The hydrophobic residues of MTHFD1L are colored in yellow
2.6 Accumulation of reactive oxygen species (ROS) mediates the antitumor activityThe accumulation of ROS, the metabolite products of mitochondria, is strongly associated with the regulation of tumor cell death [23]. Elevated levels of ROS can cause irreversible damage to cellular components, leading to apoptosis and cell death. ROS overproduction serves as an important mediator of cell death in response to various stimuli. Li Hao et al. have reported that knockdown of MTHFD1L increased ROS levels and subsequently accelerated tumor cell death under oxidative stress conditions [19]. We sought to investigate the consequences of MTHFD1L inhibition by PAA in HeLa cells. The ROS level were measured in both untreated and PAA-treated HeLa cells, with the level in control group normalized to 100%. Results demonstrated a concentration-dependent increase in ROS levels upon PAA treatment (Fig. 6A).
Fig. 6
A Generation of ROS after treatment with different concentrations of PAA in HeLa cells. B Cell viability after treatment with different concentrations of PAA in the absence or presence of ROS scavenger NAC
To further elucidate the role of ROS production in PAA-induced tumor cell death, we employed the ROS scavenger N-acetylcysteine (NAC) to assess its impact on HeLa cell viability. Our results demonstrated that pre-treatment of HeLa cells with NAC (5 mM) significantly restored cell viability across various concentrations of PAA (Fig. 6B). This finding indicates that the antitumor activity of PAA is compromised by the ROS inhibitor NAC. These data suggest that PAA stimulates the accumulation of ROS, which serves as a critical mediator of PAA-induced tumor cell death.
2.7 Antitumor activity of MTHFD1L in HeLa cellsThe MTHFD1L knockdown assay was performed to explore the functional relevance of MTHFD1L in the antitumor activity. HeLa cells were transfected with different siRNAs against MTHFD1L.The knockdown efficiency was assessed using quantitative real-time polymerase chain reaction (qRT-PCR) (Figure S3A). Our results demonstrated that cell viability was significantly reduced following MTHFD1L knockdown in HeLa cells (Figure S3B), consistent with previous reports showing that siRNA-mediated knockdown of MTHFD1L inhibits cell proliferation in colorectal cancer and papillary thyroid cancer [18, 24]. This observation limits our ability to evaluate the antitumor activity of PAA in the MTHFD1L knockdown cells.
2.8 Integrated transcriptome and network pharmacology analysisWe used the RNA sequencing to assess the overall effect of PAA on the gene expressions in HeLa cells. Analysis (Fig. 7A) revealed that 442 genes were up-regulated and 580 genes were down-regulated, respectively (Table S3). Gene ontology (GO) biological process analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis showed that PAA affected a wide range of biological functions, mainly including protein folding/refolding, response to unfolded protein/topologically incorrect protein, heat shock protein binding, providing evidences that PAA potentially targets multiple targets, including heat shock protein 90 (Fig. 7B). This is consistent with our previous report that Hsp90 is one of the main binding targets of PAA. The biosynthesis of amino acids, tyrosine metabolism, glycine, serine and threonine metabolisms were the significantly enriched pathways, supporting that PAA regulates the 1C unit metabolism.
Fig. 7
Transcriptome analysis of HeLa cells after treatment with PAA. A Volcano plot of differential gene expression associated with PAA treatment, with the horizontal axis showing the gene fold change and vertical axis indicating the p value. The red and blue circles indicate the up- and down- regulated genes, respectively. B Functional categorization of up- and down- regulated genes based on the Gene ontology (GO) annotations
Comments (0)