3.1 General experimental procedures
Optical rotations were measured on an Anton Paar MCP200 modular circular polarimeter. UV and ECD spectra were recorded on an Applied Photophysics Chirascan spectrometer. IR spectra were obtained from a PerkinElmer Spectrum Two FTIR spectrometer with a UATR accessory. 1D and 2D NMR spectra were collected from Bruker Ascend TM 500 and Bruker Avance III 400 spectrometers at 25 °C with TMS as the internal standard. HR-ESI-MS data were acquired via a Waters Micromass Q-TOF spectrometer and a X500R QTOF spectrometer from SCIEX. Semipreparative HPLC was carried out on a Shimadzu LC-20 AT equipped with an SPD-M20A PDA detector. A NanoChrom ChromCoreTM 5–120 C18 column (250 × 10 mm, 5 µm), a Phenomenex Lux cellulose-2 chiral-phase column (250 × 10 mm, 5 μm), and a YMC-pack ODS-A column (250 × 10 mm, S-5 μm, 12 nm) were utilized for HPLC purification. Solvents MeCN for HPLC were purchased from BCR International Trading Co. Ltd. Reversed-phase C18 (Rp-C18) silica gel from YMC Co. Ltd. (12 nm, S-50 μm), MCI gel from Mitsubishi Chemical Industries Ltd. (CHP20P, 75–150 μm), Sephadex LH-20 gel from Amersham Biosciences, and silica gel from Qingdao Haiyang Chemical Co, Ltd. (100–200, 300–400 mesh) were utilized for general column chromatographic separation, and fractions were monitored via silica gel TLC (GF254 plates, 0.25 mm thickness) visualized with 15% sulfuric acid in EtOH.
3.2 Fungal material
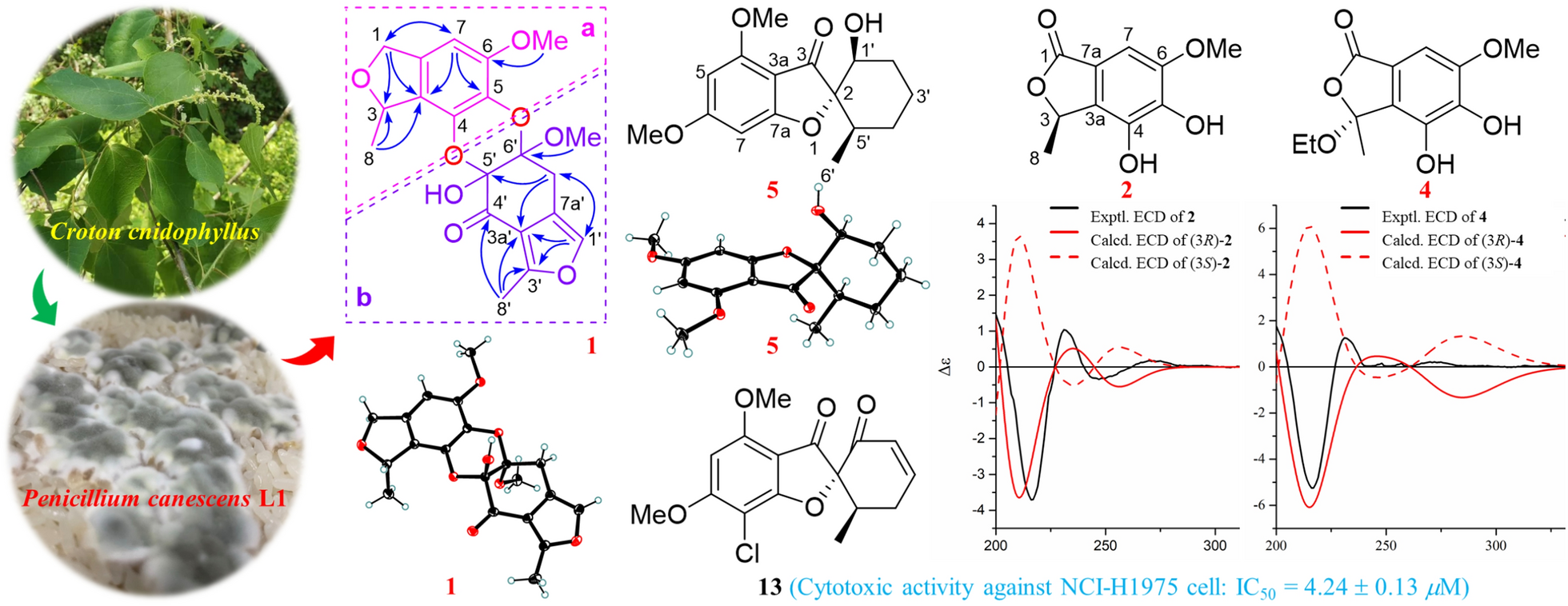
The fungal strain L1 was isolated from the fresh roots sample of Croton cnidophyllus and identified as Penicillium canescens on the base of ITS region (GenBank OQ438055). The fungal strain (No. L1) is deposited in School of Pharmaceutical Sciences, Sun Yat-sen University.
3.3 Fermentation and extraction
The strain P. canescens L1 was cultured on potato dextrose agar (PDA) plates (PDB 24.0 g and agar 18.0 g in 1.0 L H2O) at 28 °C for 7 days. The seed medium (PDB media 24.0 g in 1.0 L H2O) was inoculated with strain P. canescens L1 and incubated at 28.0 °C for 3 days on a rotating shaker (180 rpm). For chemical investigations, a large-scale fermentation of P. canescens L1 was incubated for 32 days at 28 °C in 1.5 L × 160 Erlenmeyar flasks (each flask contained 100 g rice and 100 mL H2O). After incubation, every flask was ultrasonically extracted with 4 × 0.4 L 95% EtOH for 30 min. After removing the solvents under vacuum, 250 g of crude extract was obtained, which was then suspended in water (3 L) and successively partitioned with petroleum ether (PE, 2 × 3 L) and EtOAc (3 × 3 L).
3.4 Isolation and purification
The obtained EtOAc fraction (150 g) was firstly separated over a silica gel column eluted with a gradient of PE/EtOAc (50:1 → 1:1) followed by the solvents of DCM/MeOH (70:1 → 0:1) to afford eight fractions (Frs. 1–8).
Fr. 4 (4 g) was separated by Rp-C18 silica gel CC (MeOH/H2O, 20% → 100%) to give five fractions (Frs. 4A–4E). Fr. 4A (1.3 g) was subjected to Sephadex LH-20 CC (MeOH) to yield Fr. 4A1, Fr. 4A2 (150 mg), and compound 9 (87 mg). Compound 3 (22 mg, tR 20 min) was obtained from Fr. 4A2 by semipreparative HPLC equipped with a YMC-pack ODS-A column (MeCN/H2O, 27:73, 3 mL/min). Compound 29 (23 mg) was precipitated from Fr. 4E (160 mg).
Fr. 5 (18.4 g) was subjected to Rp-C18 silica gel CC (MeOH/H2O, 20% → 65%) to give five fractions (Frs. 5A–5E). Fr. 5A was divided into three fractions (Frs. 5A1–5A3) by Sephadex LH-20 CC (MeOH). Fr. 5A2 (4.6 g) was further divided into seven fractions (Frs. 5A2A–5A2G) by silica gel CC (PE/DCM, 1:1 → 0:1 and then DCM/MeOH, 500:1 → 70:1). Fr. 5A2D (661 mg) was separated by Sephadex LH-20 CC (MeOH) to give three fractions (Frs. 5A2D1–5A2D3). Compound 20 (8 mg, tR 10 min) was obtained from Fr. 5A2D1 (17 mg) by semipreparative HPLC equipped with a chiral column (MeCN/H2O, 45:55, 3 mL/min), 19 (58 mg, tR 8 min) was obtained from Fr. 5A2D2 (219 mg) by semipreparative chiral HPLC (MeCN/H2O, 40:60, 3 mL/min), and 18 (19 mg, tR 8 min) was obtained from Fr. 5A2D3 (270 mg) by semipreparative chiral HPLC (MeCN/H2O, 35:65, 3 mL/min). Fr. 5B (824 mg) was separated by silica gel CC (PE/DCM, 1:1 → 0:1 and then DCM/MeOH, 500:1 → 70:1) to give Frs. 5B1–5B4 and compound 17 (10 mg). Compounds 4 (18 mg) and 2 (47 mg) were obtained from Fr. 5B1 (90 mg) by Sephadex LH-20 CC (MeOH). Fr. 5C (934 mg) was further divided into six fractions (Frs. 5C1–5C6) by silica gel CC (PE/DCM, 1:1 → 0:1 and then DCM/MeOH, 500:1 → 70:1). Fr.5C2 (260 mg) was successively purified by Sephadex LH-20 CC (MeOH) and semipreparative HPLC equipped with a NanoChrom ChromCoreTM 5–120 C18 column (MeCN/H2O, 30:70, 3 mL/min) to yield 6 (44 mg, tR 7.5 min). Fr. 5E (3.9 g) was separated by Sephadex LH-20 CC (MeOH) to give three fractions (Frs. 5E1–5E3). Fr. 5E1 (1.93 g) was further divided into three fractions (Frs. 5E1A–5E1C) by silica gel CC (PE/DCM, 1:1 → 0:1 and then DCM/MeOH, 500:1 → 70:1). Compounds 11 (83 mg, tR 7.5 min) and 13 (24 mg, tR 10.5 min) were obtained from Fr. 5E1A (200 mg) by semipreparative HPLC equipped with a NanoChrom ChromCoreTM 5–120 C18 column (MeCN/H2O, 60:40, 3 mL/min). Fr. 5E1B (520 mg) was subjected to Sephadex LH-20 CC (MeOH) and followed by semipreparative chiral HPLC (MeCN/H2O, 53:47, 3 mL/min) to give 5 (14 mg, tR 10 min) and 10 (64 mg, tR 12.5 min). Fr. 5E1C (818 mg) was subjected to silica gel CC (PE/EtOAc, 70:1 → 0:1) to yield 24 (20 mg) and 1 (2 mg). Fr. 5E2 (1.2 g) was separated by silica gel CC (PE/EtOAc, 70:1 → 1:1) to give compound 22 (220 mg) and Frs. 5E2A–5E2E. Fr. 5E2E (63 mg) was further purified by semipreparative chiral HPLC (MeCN/H2O, 50:50, 3 mL/min) to yield 21 (30 mg, tR 6 min) and 23 (2 mg, tR 8 min).
Fr. 6 (24 g) was subjected to Rp-C18 silica gel CC (MeOH/H2O, 20% → 70%) to give six fractions (Frs. 6A–6F). Fr. 6A (5.2 g) was further separated by silica gel CC (DCM/MeOH, 500:1 → 70:1) to yield Frs. 6A1–6A5, compound 8 (84 mg), and Fr. 6A6 (300 mg). Fr. 6A6 was firstly separated by semipreparative chiral HPLC (MeCN/H2O, 15:85, 3 mL/min) to give two fractions (Frs. 6A7A–6A7B). Then, Fr. 6A7A (91 mg) was purified by HPLC equipped with a NanoChrom ChromCoreTM 5–120 C18 column (MeCN/H2O, 15:85, 3 mL/min) to yield 16 (21 mg, tR 9 min), and compound 7 (8 mg, tR 8 min) was obtained from Fr. 6A7B (40 mg) also by HPLC equipped with a NanoChrom ChromCoreTM 5–120 C18 column (MeCN/H2O, 20:80, 3 mL/min). Fr. 6E (4.7 g) was further separated by silica gel CC (DCM/MeOH, 500:1 → 70:1) to give Frs. 6E1–6E9, 28 (23 mg), and 34 (14 mg). Fr. 6E2 (200 mg) was purified by semipreparative HPLC equipped with a YMC-pack ODS-A column (MeCN/H2O, 45:55, 3 mL/min) to yield 12 (22 mg, tR 20 min), 15 (3 mg, tR 21 min), and 14 (14 mg, tR 23.5 min). Fr. 6E5 (510 mg) was separated by Sephadex LH-20 CC (MeOH) to give 25 (34 mg). Fr. 6E6 (500 mg) was subjected to Sephadex LH-20 CC (MeOH) to yield two fractions (Frs. 6E6A–6E6B). Fr. 6E6B (250 mg) was further purified over semipreparative HPLC equipped with a YMC-pack ODS-A column (MeCN/H2O, 35:65, 3 mL/min) to afford 31 (3 mg, tR 17 min) and 33 (1 mg, tR 20 min). Fr. 6E8 (220 mg) was divided into compound 30 (20 mg) and Fr. 6E8A (150 mg) by Sephadex LH-20 CC (MeOH), and then compound 32 (16 mg, tR 17 min) was obtained from Fr. 6E8A by HPLC equipped with a NanoChrom ChromCoreTM 5–120 C18 column (MeCN/H2O, 40:60, 3 mL/min). Fr. 6F (800 mg) was subjected to silica gel CC (PE/DCM, 1:1 → 0:1 and then DCM/MeOH, 500:1 → 70:1) to give compound 26 (140 mg) and Fr. 6F1 (150 mg). Compound 27 (8 mg, tR 16 min) was obtained from Fr. 6F1 by semipreparative HPLC equipped with a YMC-pack ODS-A column (MeCN/H2O, 68:32, 3 mL/min).
3.5 Spectroscopic data of compounds3.5.1 Penicanesol A (
1)
Colorless needles; m.p. 256–257; \([\alpha ]_}}^}}\) + 19.9 (ca. 0.09, MeCN); UV (MeCN) λmax (log ε) 230 (3.50) nm; ECD (ca. 2.6 × 10−4 M, MeCN) λmax (Δε) 203 (+4.50), 218 (−2.06), 240 (+ 0.26) nm; IR (UATR) νmax 3357, 2921, 2852, 1705, 1612, 1572, 1496, 1349, 1130 cm−1; 1H and 13C NMR data see Table 1; HR-ESI-MS m/z 411.1055 [M + Na]+ (calcd for C20H20O8Na+, 411.1050).
3.5.2 Penicanesol B (
2)
White amorphous solid; \([\alpha ]_}}^}}\) + 36.9 (ca. 0.12, MeCN); UV (MeCN) λmax (log ε) 190 (3.50) nm; ECD (ca. 4.8 × 10−4 M, MeCN) λmax (Δε) 193 (+1.84), 216 (−3.70), 231 (+ 1.04), 247 (−0.35) nm; IR (UATR) νmax 3349, 1720, 1630, 1492, 1447, 1353, 1113 cm−1; 1H and 13C NMR data see Table 2; HR-ESI-MS m/z 233.0422 [M + Na]+ (calcd for C10H10O5Na+, 233.0420).
3.5.3 Penicanesol C (
3)
White amorphous solid; \([\alpha ]_}}^}}\) + 12.5 (c 0.13, MeCN); UV (MeCN) λmax (log ε) 190 (3.70) nm; ECD (ca. 4.5 × 10−4 M, MeCN) λmax (Δε) 193 (+1.96), 216 (−4.63), 231 (+ 1.11), 247 (−0.37) nm; IR (UATR) νmax 3353, 1645, 1516, 1489, 1043 cm−1; 1H and 13C NMR data see Table 2; HR-ESI-MS m/z 225.0743 [M + H]+ (calcd for C11H13O5+, 225.0757), 247.0565 [M + Na]+ (calcd for C11H12O5Na+, 247.0577).
3.5.4 Penicanesol D (
4)
White amorphous solid; \([\alpha ]_}}^}}\) + 44.9 (c 0.10, MeCN); UV (MeCN) λmax (log ε) 210 (4.80) nm; ECD (ca. 3.9 × 10−4 M, MeCN) λmax (Δε) 193 (+2.22), 216 (−5.25), 231 (+ 1.25) nm; IR (UATR) νmax 3361, 2921, 2851, 1738, 1629, 1497, 1462, 1184 cm−1; 1H and 13C NMR data see Table 2; HR-ESI-MS m/z 277.0687 [M + Na]+ (calcd for C12H14O6Na+, 277.0683).
3.5.5 Penicanesol E (
5)
Colorless needles; \([\alpha ]_}}^}}\) + 58.9 (ca. 0.11, MeCN); UV (MeCN) λmax (log ε) 254 (4.70) nm; ECD (ca. 3.4 × 10−4 M, MeCN) λmax (Δε) 192 (−12.06), 215 (+ 15.63), 233 (−3.04), 280 (+ 2.60), 329 (−4.75) nm; IR (UATR) νmax 3479, 1681, 1615, 1596, 1459, 1216, 1155 cm−1; 1H and 13C NMR data see Table 3; HR-ESI-MS m/z 315.1206 [M + Na]+ (calcd for C16H20O5Na+, 312.1203).
3.5.6 Penicanesol F (
6)
White amorphous solid; UV (MeCN) λmax (log ε) 254 (4.81) nm; 1H and 13C NMR data see Table 3; HR-ESI-MS m/z 277.0678 [M + Na]+ (calcd for C12H14O6Na+, 277.0683).
3.5.7 Penicanesol G (
7)
White amorphous solid; \([\alpha ]_}}^}}\) + 34.8 (ca. 0.14, MeCN); UV (MeCN) λmax (log ε) 216 (3.49), 238 (2.50), 266 (2.98) nm; ECD (ca. 5.1 × 10−4 M, MeCN) λmax (Δε) 193 (+1.71), 216 (−4.05), 231 (+ 0.97), 247 (−0.32) nm; IR (UATR) νmax 3275, 1702, 1621, 1522, 1497, 1317, 1043 cm−1; 1H and 13C NMR data see Table S2.1 in Supporting Information; HR-ESI-MS m/z 197.0441 [M + H]+ (calcd for C12H15O6+, 197.0444).
3.6 Crystallographic data
Compounds 1, 5, 8, and 13 were recrystallized MeOH/DCM (5:1) to afford colorless needles at room temperature. Crystallographic data for the structures determined in this study have been deposited at the Cambridge Crystallographic Data Centre (deposition number: 2402573 for 1, 2402743 for 5, 2407293 for 8, and 2402745 for 13) and can be obtained free of charge from the CCDC Web site (https://www.ccdc.cam.ac.uk/).
Penicanesol A (1): Crystal Data for C20H20O8 (M = 388.36 g/mol): orthorhombic, space group P212121 (no. 19), a = 8.79670(9) Å, b = 12.20777(15) Å, c = 16.4867(2) Å, V = 1770.47(4) Å3, Z = 4, T = 100.00(10) K, μ(Cu Kα) = 0.959 mm−1, Dcalc = 1.457 g/cm3, 18,304 reflections measured (9.014° ≤ 2θ ≤ 158.02°), 3748 unique (Rint = 0.0477, Rsigma = 0.0310) which were used in all calculations. The final R1 was 0.0372 (I > 2σ(I)) and wR2 was 0.1020 (all data). Flack parameter = 0.02(8).
Penicanesol E (5): Crystal Data for C16H20O5 (M = 292.32 g/mol): monoclinic, space group P21 (no. 4), a = 6.08950(10) Å, b = 10.3226(2) Å, c = 11.9172(2) Å, β = 104.211(2)°, V = 726.18(2) Å3, Z = 2, T = 99.99(10) K, μ(Cu Kα) = 0.818 mm−1, Dcalc = 1.337 g/cm3, 7450 reflections measured (7.652° ≤ 2Θ ≤ 156.932°), 2703 unique (Rint = 0.0154, Rsigma = 0.0132) which were used in all calculations. The final R1 was 0.0270 (I > 2σ(I)) and wR2 was 0.0716 (all data). Flack parameter = 0.09 (4).
Penicanesin E (8): Crystal Data for C10H10O6 (M = 226.18 g/mol): monoclinic, space group P21/c (no. 14), a = 8.30853(9) Å, b = 14.86270(18) Å, c = 7.72506(8) Å, β = 97.8579(10)°, V = 944.989(18) Å3, Z = 4, T = 100.00(10) K, μ(Cu Kα) = 1.155 mm−1, Dcalc = 1.590 g/cm3, 18,746 reflections measured (10.75° ≤ 2Θ ≤ 157.508°), 2007 unique (Rint = 0.0443, Rsigma = 0.0214) which were used in all calculations. The final R1 was 0.0365 (I > 2σ(I)) and wR2 was 0.0983 (all data).
4′-Demethoxyisogriseofulvin (13): Crystal Data for C16H15ClO5 (M = 322.73 g/mol): orthorhombic, space group P212121 (no. 19), a = 11.3451(2) Å, b = 16.9711(3) Å, c = 38.8580(6) Å, V = 7481.7(2) Å3, Z = 20, T = 100.00(10) K, μ(Cu Kα) = 2.462 mm−1, Dcalc = 1.433 g/cm3, 73,432 reflections measured (4.548° ≤ 2Θ ≤ 157.614°), 15,741 unique (Rint = 0.0862, Rsigma = 0.0645) which were used in all calculations. The final R1 was 0.0948 (I > 2σ(I)) and wR2 was 0.2488 (all data). Flack parameter = 0.034 (7). Flack parameter = 0.09 (4).
3.7 ECD calculations
Please see S1.1 in Supporting Information for details of the quantum chemical ECD calculation of 2 and 4.
3.8 Cytotoxic activity assay
For cell viability, NCI-H1975 cells were seeded in 96-well plates at 2,000 cells per well (optimum density for growth) in a total volume of 100 μL of media containing 10% serum. Serially diluted compounds in 50 μL of media were added to the cells 24 h later. After 3 days of incubation, Cell Counting Kit-8 reagents (Dojindo, Japan) were added, and luminescence was measured according to the manufacturer’s instructions. All experiments were repeated three times. The data are presented as percentage of viable cells with vehicle-treated cells set as 100. The estimated in vitro IC50 values were calculated by using GraphPad Prism 9 software.




Comments (0)