
Remember me



Compound 1 was obtained as a yellow oil with the molecular formula C27H33NO6 determined by HRESIMS at m/z 468.2383 [M + H]+ (calcd. for C27H34NO6+, 468.2381), corresponding to 12 degrees of unsaturation. The infrared absorption spectrum (IR) peaks at 3443.9, 1683.3, and 1628.6 cm−1 correspond to the stretching vibrations of the hydroxyl, carboxylic carbonyl, and amide carbonyl groups, respectively. Its ultraviolet (UV) spectrum exhibited an absorption maximum at 257 nm, indicating the existence of conjugated systems.
The 1H NMR data (Table 1) revealed characteristic signals for a monosubstituted phenyl at δH 7.11 (2H, d, J = 7.6 Hz, H-22/H-26), 7.31 (2H, t, J = 7.6 Hz, H-23/H-25), and 7.24 (1H, t, J = 7.6 Hz, H-24), three singlet methyl groups at δH 1.40 (3H, s, H3-11), 1.64 (3H, s, H3-12), and 2.17 (3H, s, H3-28), a doublet methyl group at δH 1.16 (3H, d, J = 7.0 Hz, H3-20), and one amide NH proton at δH 6.09. The spectroscopic data also indicated two distinct groups of isolated and geminally coupled protons, one methylene at δH 2.61 (1H, dd, J = 13.3, 7.5 Hz) and 2.76 (1H, dd, J = 13.3, 7.8 Hz), the other at δH 1.65 (1H, m) and 1.47 (1H, ddd, J = 14.4, 8.7, 6.0 Hz). The 13C NMR, DEPT, and HSQC data of 1 displayed 27 carbon signals, including a carboxylic acid carbon at δC 179.6 (C-19), an amide carbonyl at δC 177.4 (C-1), an ester carbonyl at δC 170.0 (C-27), a double bond at δC 127.3 (C-5) and 134.8 (C-6), one nonprotonated carbon at δC 63.3 (C-9), two methylene carbons at δC 36.3 (C-17) and 42.7 (C-10), nine sp3 methine carbons at δC 28.9 (C-13), 29.8 (C-14), 32.0 (C-15), 38.6 (C-18), 47.5 (C-4), 52.4 (C-8), 58.1 (C-3), 70.8 (C-7), and 78.1 (C-16), two of which are oxygenated. These findings suggest that compound 1 contains four double bonds and one phenyl group, inferring the presence of four rings based on the 12 degrees of unsaturation derived from the molecular formula.
Table 1 The 1H NMR data (600 MHz) and 13C NMR data (150 MHz) of compounds 1 and 2 in CDCl3 (δ in ppm)The 1H–1H COSY spectrum of 1 indicated three proton spin–spin systems, namely H2-10/H-3/H-4/2-NH, H-7/H-8/H-13/H-14(/H-15/H-16)/H2-17/H-18/H3-20, and H-22/H-23/H-24/H-25/H-26 (Fig. 2). The HMBC correlations (Fig. 2) between NH and C-1/C-3/C-4/C-9, H-4 and C-1/C-6/C-10, H-7 and C-5, H3-11 and C-4/C-5/C-6, H3-12 and C-5/C-6/C-7, as well as H-8 and C-4/C-6 established the structural connectivity between the five-membered lactam ring (A ring) and the six-membered ring (B ring). A proposed ring C fused with a tricyclic ring D was deduced by the HMBC correlations between H-13 and C-7/C-16/C-17, H-15 and C-9/C-8/C-17, H-16 and C-1/C-8/C-14, as well as H-14 and C-8/C-16, along with the 1H–1H COSY correlations of H-13/H-15/H-14. Additionally, the spin–spin coupling of H-14/H2-17/H-18/H3-20, and the HMBC correlations between H3-20 and C-17/C-18/C-19, H2-17 and C-13/C-15/C-19 suggested the presence of a 2-methylpropanoic acid moiety attached to C-14 of the tricyclic ring (D ring). Further HMBC correlation between H-16 and C-27 verified the attachment of the acetyl group at C-16. Thus, the planar structure of compound 1 was defined as a novel cytochalasan skeleton with a unique bicyclo[3.1.0]hexane moiety.
Fig. 2
1H–.1H COSY and key HMBC correlations of compounds 1 and 2
Nuclear Overhauser effects (NOEs) between H-4 and H-16, as well as between H2-10 and H-16, suggest that these protons are on the same side of the five-membered lactam ring. The NOESY spectrum showed correlations of H-4/H-8/H-14 and of H-7/H-13, suggesting that H-4, H-8, and H-14 are cofacial, while H-7 and H-13 are cofacial (Fig. 3). The equatorial position of H-7 was inferred from the coupling constant between H-7 and H-8 (J = 10.5 Hz) (Table 1). Previous studies have indicated that the essential elements of most cytochalasan skeletons share the same stereochemistry, with configurations at C-3, C-4, C-7, C-8, C-9, and C-16 assigned as 3S, 4R, 7S, 8R, 9R, and 16R, respectively [5, 13], which is consistent with the aforementioned NOE analyses. However, the relative configuration of ring D could not be fully elucidated by NOESY experiments. Fortunately, coupling constants between vicinal protons in ring D provide valuable information for determining their relative orientations. H-13 exhibited coupling constants of 7.7, 4.2, and 3.8 Hz with H-15, H-8, and H-14, respectively. The small coupling constant between H-13 and H-14 (J = 3.8 Hz) and the large coupling constant between H-13 and H-15 (J = 7.7 Hz) suggest that H-13 and H-15 are coplanar and both diaxial to H-14.
Fig. 3
Key NOE correlations of compounds 1 and 2, depicted with their optimal conformations (coordinates are provided in Tables S14 and S22)
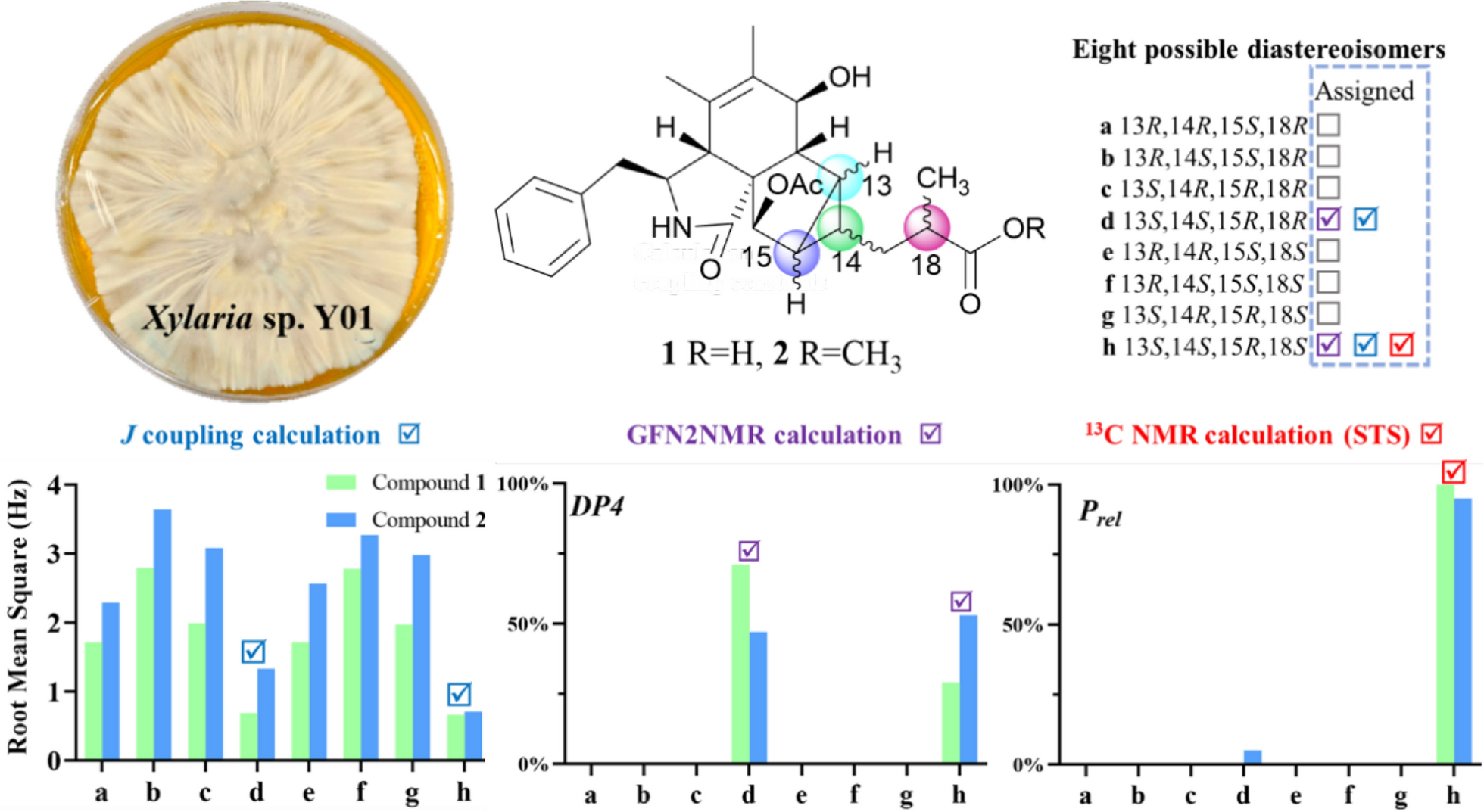
To confirm the relative configuration of ring D, the J coupling constants for different possible conformations of ring D were calculated [14]. The fusion of rings C and D is restricted to a cis configuration due to strain that inhibits the trans configuration from achieving a stable geometric structure. Therefore, the relative configuration of 1 is one of the eight possible relative diastereoisomers (1a–1h), as shown in Fig. 4. The J coupling constants of protons on rigid rings for these eight possible epimers were correlated with the experimental coupling constants to calculate the mean absolute error (MAE) and root mean square error (RMS). The results indicate that the relative configurations of 1d and 1h align more closely with the experimental values, as they exhibit lower MAE and RMS compared to the other epimers (Table 2). These findings are consistent with the previous analysis.
Fig. 4
Eight possible epimers of compounds 1 and 2
Table 2 Experimental J coupling constants (CDCl3) for compounds 1 and 2, and theoretical values for eight possible epimers (detailed data are provided in Table S5 and S6; the values in bold font are the optimal values in their respective columns)To discern the chirality at key positions from an alternative perspective, 13C NMR calculations were utilized by analyzing chemical shift differences. The GFN2NMR method, a semiempirical approach integrated into a deep graph convolutional network, facilitates rapid and precise 13C NMR chemical shift calculations, noted for its high accuracy and low computational cost [15]. It is capable of distinguishing diastereomers based on the rules learned from large datasets. The most likely structures of 1 were swiftly identified from eight possible isomers using the GFN2NMR method. Since their Pmean values fall within the confidence interval and Prel exceeds 5% (1d at 71%, 1h at 29%), both 1d and 1h are assigned as plausible configurations (Table 3).
Table 3 Experimental 13C NMR chemical shifts (CDCl3) of compounds 1 and 2, and the calculated chemical shifts for the eight possible epimers predicted by GFN2NMR (detailed data are provided in Table S1 and S2; the values in bold font are the optimal values in their respective columns)The GIAO 13C NMR calculations with the sorted training sets (STS) protocol showed significantly enhanced accuracy and reliability for structural determination [16] (e. g., pestalopyrones A–D [12]), which provided a tool based on quantum chemistry. The calculated chemical shifts by STS for the eight possible epimers 1a–1h were correlated with the experimental 13C NMR chemical shifts to determine MAE, RMS, and the statistical parameters Pmean and Prel (Table 4). The 13C NMR calculations for compound 1h closely matched the experimental data, with both the MAE and RMS values significantly lower than those for 1b. Furthermore, the Prel probability calculations assigned a 100% likelihood to 1h. This result not only established the chirality at C-18 but also validated the conformation of the D ring, consistent with the possible structures inferred from J coupling constants calculations and GFN2NMR analysis. An ECD calculation for 1h was conducted to determine the absolute configuration of 1, indicating a configuration of 3S,4R,7S,8R,9R,13S,14S,15R,16R,18S, which matched well with the experimental ECD data as shown in Fig. 5. These findings confirm that 1h is the correct structure of xylariaide A.
Table 4 13C NMR chemical shifts calculation (CDCl3) of structures a–h fitting to the experimental data of compounds 1 and 2 using GIAO 13C NMR calculations with the STS protocol (detailed data are provided in Table S3 and S4; the values in bold font are the optimal values in their respective columns)Fig. 5
The experimental and calculated ECD curves of 1 and 2 and their enantiomers
Compound 2 was obtained as an amorphous powder, with a molecular formula of C28H35NO6, as determined by HRESIMS at m/z 482.2538 [M + H]+ (calcd. for C28H36NO6+, 482.2538). The one-dimensional spectrum data of compound 2 exhibited an additional methoxyl group at δH 3.68 (3H, s) and δC 51.7, with all other signals closely resembling those of 1 except for the active proton signal of 2-NH (Table 1). The HMBC correlation between -OCH3 and C-19 confirmed the attachment of the methoxyl group at C-19. NOE signals indicate that H-4, H2-10, and H-16 are on the same side of the five-membered lactam ring, while H3-11 and H-3 are on the opposite side. The optimal conformations of compounds 1 and 2, as illustrated in Fig. 3, differ significantly in the configuration of the 2-methylpropionic acid moiety, leading to the absence of a distinct NOE signal between H-14 and H-18 for 2. The remaining NOE signals for 2 are consistent with those of 1. Similarly, the J coupling constants and 13C NMR calculations of 2 were also performed, aligning with the results for 1 and confirming the relative configuration of 2. The experimental ECD spectrum of 2 displayed two positive Cotton effects at 215 nm (Δε + 52.6) and 258 nm (Δε + 3.5), similar to the spectrum of 1. Absolute confirmation was determined by ECD calculation, with the experimental values closely matching those for the configuration 3S,4R,7S,8R,9R,13S,14S,15R,16R,18S, namely xylariaide B.
Upon comparing the structures of compounds 1 and 2, it is evident that their only difference lies in the methoxy substitution on the 19-COOH group at the side chain. In the 1D NMR spectra, most of their signals are remarkably similar. However, the chemical shift of the 2-NH proton, which is distant from the side chain, shows a significant difference of 0.6 ppm. To investigate the cause of this discrepancy and further validate the structural assignments, we analyzed the low-energy conformations of compounds 1 and 2, as shown in Fig. 6. It revealed that the most dominant conformation of compound 1 features the 10-phenyl ring and the N-2 group adopting an anti-conformation. In contrast, for compound 2, the gauche conformation is energetically more favorable and predominant. In this conformation, the 2-NH proton lies within the shielding region of the 10-phenyl ring, which explains why the chemical shift of 2-NH in compound 2 appears at a higher field in the 1H NMR spectrum. Further examination of their geometric structures revealed that in the lowest-energy conformation of compound 1, a weak hydrogen bond (bond length: 2.28 Å) forms between the 19-COOH and 27-CO groups. This interaction pulls the 16-acetyl group away from the 10-phenyl ring (the distance between 22-H and C-27 is 2.79 Å, and the dihedral angle between C-27 and H-16 is 33.5º), allowing the phenyl ring to adopt the energetically favorable anti-conformation. In contrast, in the anti-conformation of compound 2, the 16-acetyl group is closer to the 10-phenyl ring (the distance between 22-H and C-27 is 2.66 Å, and the dihedral angle between C-27 and H-16 is 22.3º). The resulting steric hindrance likely increases the energy of the anti-conformation, making the gauche conformation the lowest-energy state. This analysis not only provides a clear explanation for why a simple methoxy substitution on the 19-COOH group can lead to a significant change in the chemical shift of the distant 2-NH proton, but also further corroborates the validity of the structural assignments.
Fig. 6
Analysis of the magnetic anisotropic effect of the benzene ring on the 2-NH proton in compounds 1 and 2. Conformer 1 of compounds 1 and 2 represents the global minimum conformation, respectively
2.2 Proposed biosynthetic pathwayCompounds 1 and 2 contain the same rings A and B as curtachalasin J [17], a co-isolated known cytochalasan identified from this strain (NMR data provided in Figs. S22 and S23), and also feature a benzyl group at C-3. This observation suggests that the 5/6/5/3 ring system common to compounds 1 and 2 may be biogenetically derived from the known compound curtachalasin J. Intramolecular cyclization between C-13 and C-20 in curtachalasin J could result in the bicyclo[3.1.0]hexane moiety. Besides, inspired by the C–C bond cleavage reactions on α-hydroxyketones [18], we propose that the carbon bonds between C-17 and C-18 and between C-17 and C-19 within ring E undergo oxidative cleavage, and both C-17 and C-19 are transformed into carboxylic acids. The following decarboxylation at C-19 leads to the formation of compound 1. The stereochemistry of C-8, C-13, C-14, C-16, C-20, and C-21 in curtachalasin J corresponds to that of C-8, C-13, C-14, C-18, C-15, and C-16 in compound 1, respectively, further supporting the proposed biogenetic pathway (Scheme 1).
Scheme 1
Proposed Biosynthetic Pathway of compounds 1 and 2
2.3 Preliminary bioactivity screening resultsThe cytochalasans, a well-known family of fungal metabolites, are recognized for their ability to inhibit the polymerization of filamentous (F)-actin [19]. These compounds have shown significant anticancer properties in numerous in vitro and in vivo studies [20]. To evaluate the potential anticancer properties of compounds 1 and 2, a cytotoxicity assessment was conducted on three cancer cell lines (HCT116, 4T1, and MHCC97H) and one normal cell line (GES-1). Compounds 1 and 2 did not exhibit significant cytotoxic effects against any of the cell lines tested.
To assess the antimicrobial activity of compounds 1 and 2, a screen for antibacterial activity against Bacillus subtilis, Escherichia coli, Staphylococcus aureus, Acinetobacter baumannii, and Pseudomonas aeruginosa, as well as for antifungal activity against Saccharomyces cerevisiae and Canidia albicans, was conducted. Both compounds showed MIC values above 50 μM, indicating no significant antimicrobial activity against the bacterial strains tested.
The anti-inflammatory activity of compounds 1 and 2 was evaluated by assessing their inhibitory effects on nitric oxide (NO) production in lipopolysaccharide (LPS)-stimulated RAW264.7 macrophages. Compounds 1 and 2 demonstrated no significant inhibitory activity on NO production at a concentration of 30 μM.
Comments (0)