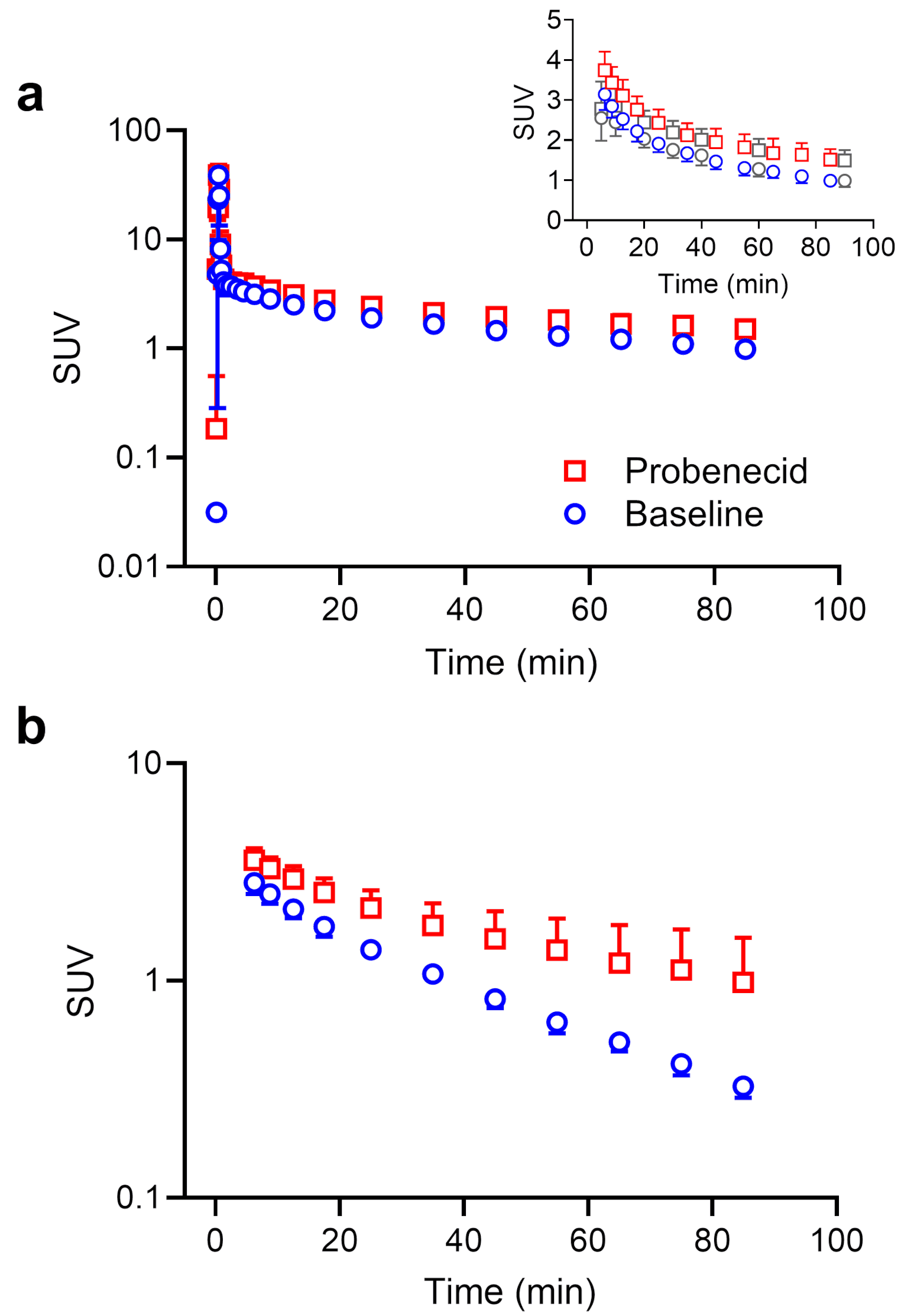
We found significantly decreased elimination of [11C]BMP-derived radioactivity from several MRP-expressing tissues following probenecid administration, supporting the suitability of [11C]BMP to measure MRP function in humans with LAFOV PET/CT at a whole-body, multi-tissue level.
In previous rodent studies with [11C]BMP, kE has been proposed as a parameter for tissue MRP1 function, which can be directly derived from the tissue TACs without the need to consider plasma radioactivity [10,11,12]. kE values in the brain and lungs of Abcc1(−/−) mice or MRP1 inhibitor-treated wild-type mice were markedly decreased relative to untreated wild-type mice, supporting that kE can be used to estimate tissue MRP1 function [10,11,12,13]. The underlying assumption is that after i.v. injection [11C]BMP is efficiently delivered to tissues by passive diffusion followed by rapid and quantitative conversion via intracellular glutathione-S-transferases (GSTs) into its glutathione-conjugate [11C]MPG, which cannot diffuse back into blood due its high polarity and whose tissue elimination is dependent on MRP1 function [10]. At the same time, [11C]BMP is also rapidly converted in blood into [11C]MPG, which can only poorly distribute from blood into tissues. Therefore, after initial tissue delivery no exchange of [11C]BMP between blood and tissue occurs, so that the kE parameter is assumed to reflect tissue transporter function. This has been validated in rodent studies, in which chromatographic analysis of tissue samples (i.e., brain and lung) revealed rapid and almost quantitative conversion of [11C]BMP into [11C]MPG within 5–15 min after radiotracer injection [10,11,12, 14]. Moreover, after i.v. injection of radiolabelled MPG into mice there was negligible brain and lung uptake of radioactivity, supporting that the glutathione-conjugate poorly distributes from blood into tissues [11, 15]. To estimate the extent of [11C]BMP conversion into [11C]MPG in human tissues, we analysed venous plasma samples, obtained at different time points after radiotracer injection, with radio-HPLC. Indeed, the major radiolabelled species in plasma represented the radiolabelled glutathione-conjugate (66 ± 5% of total radioactivity at 5 min after radiotracer injection) (Supplementary Fig. 1). We also detected some unconverted [11C]BMP in plasma. This may represent the plasma protein-bound fraction of [11C]BMP which is not available for distribution into red blood cells where glutathione-conjugation presumably occurs [16]. Moreover, an unidentified, lipophilic radiolabelled metabolite was detected in plasma (Supplementary Fig. 1), from which it is not known whether it can distribute to different tissues. Given the rapid conversion of [11C]BMP into [11C]MPG in human blood, it can be expected that efficient conversion will also occur in different human tissues in which GSTs are ubiquitously expressed, suggesting that kE can be used in humans, like in rodents, as a parameter for tissue MRP function. To eliminate the bias of changes in blood radioactivity concentrations following probenecid administration in the calculation of kE values, we corrected the tissue TACs for vascular radioactivity content by using tissue blood volume fractions available from literature. Since probenecid does not have any vasodilatation or vasoconstriction effects, it was assumed that tissue blood volume fractions remained unchanged after probenecid administration.
To prove transport of [11C]BMP-derived radioactivity by MRP1 and possibly other MRP subtypes in human tissues, study participants underwent [11C]BMP PET scans without and with pre-treatment with an MRP inhibitor. Since MK571 which had been previously used in rodent studies [11,12,13,14] is not available for clinical application, we instead administered the prototypical organic anion transporter inhibitor probenecid, a drug prescribed for the treatment of gout and hyperuricemia. Among other organic anion transporters (organic anion transporter 1 and 3 - OAT1/SLC22A6 and OAT3/SLC22A8 -, and MRP2-5/ABCC2-5), probenecid also inhibits MRP1 in vitro [25], and in vivo in mice [26] and humans [27]. The probenecid dose employed in our study was four times higher than the clinically used dose for the treatment of hyperuricemia. In a previous study [28], a maximum unbound plasma concentration of approximately 50 µmol/L was reached at 3–4 h after oral administration of 2 g probenecid to healthy subjects. In this concentration range, MRP1 inhibition by probenecid has been observed in vitro [25].
Following probenecid administration, the elimination of [11C]BMP-derived radioactivity from plasma and its urinary excretion were significantly decreased (Figs. 1b and 3a), which pointed to an inhibition of renal transporters. The decreased urinary excretion of radioactivity after probenecid administration led to increased blood radioactivity concentrations necessitating the correction of tissue TACs for vascular radioactivity content for the calculation of kE values.
In human kidney proximal tubule cells, OAT1 and OAT3 are expressed in the basolateral (blood-facing) plasma membrane, while MRP2 and MRP4 are expressed in the apical brush-border membrane (Supplementary Fig. 5d) [3]. These transporters work together in mediating the urinary excretion of various drugs and drug metabolites [3]. Experiments in Abcc4(−/−) mice had shown that MRP4 contributes to the urinary excretion of [11C]BMP-derived radioactivity in mice [12]. Our observation that CLurine, plasma, total was reduced to a greater extent than CLurine, kidney, total (Fig. 3b) suggests that both basolateral uptake transporters (i.e., OAT3) and apical efflux transporters (MRP2, MRP4) are involved in the urinary excretion of [11C]BMP-derived radioactivity (Supplementary Fig. 5d). This is in good agreement with mouse experiments, in which MK571 inhibited both the renal uptake and efflux clearances of [11C]BMP-derived radioactivity [12]. The decrease in CLurine, plasma, [11C]MPG was greater than that of CLurine, kidney, total (Fig. 3b) indicating that [11C]MPG is the radiolabelled species which is preferentially excreted via renal organic anion transporters. In addition to the decreased urinary clearance values, kE was also significantly reduced in the kidneys (Fig. 7).
We also observed a significant kE decrease in the liver (Fig. 7). However, the amount of radioactivity excreted into bile was low (< 5% of the administered activity) and not affected by probenecid administration (Supplementary Fig. 3). The kE reduction in the liver may therefore not be due to inhibition of MRP2-mediated biliary excretion of radioactivity, but rather to inhibition of MRP3- and/or MRP4-mediated efflux of radioactivity from hepatocytes into blood (Supplementary Fig. 5e) [3].
Apart from the effects seen in excretory organs, we observed significant kE decreases in the myocardium and skeletal muscle after probenecid administration (Fig. 7). MRP1 is expressed in the sarcolemmal membrane of cardiomyocytes [29] and skeletal muscle fibres [30] (Supplementary Fig. 5b, c). In the myocardium, MRP1 was shown to protect the heart from doxorubicin-induced cardiotoxicity by direct cellular export of doxorubicin and/or by cellular export of cytotoxic products generated during doxorubicin-induced oxidative stress [29]. In skeletal muscle, MRP1 was shown to control intracellular concentration levels of lipid-lowering statin drugs and to protect muscle cells from statin-induced muscle toxicity [30].
In the lungs, MRP1 is abundantly expressed in the basolateral membrane of airway epithelial cells (Supplementary Fig. 5a) [31]. [11C]BMP has been successfully used to measure MRP1 function in the rodent lungs either after i.v. administration [11, 12] or after intratracheal aerosolisation [14]. However, in the present study no significant kE decrease was observed in the lungs following probenecid administration (Fig. 7). This may be explained by insufficiently high tissue probenecid concentration levels to achieve effective inhibition of MRP1 in airway epithelial cells.
In the brain, MRP1 is predominantly expressed in parenchymal cells (i.e., neurons and astrocytes) (Supplementary Fig. 4a) and in choroid plexus epithelial cells (Supplementary Fig. 4b) and only to a low extent in brain capillary endothelial cells [32]. It has been proposed that [11C]BMP is converted in mouse brain parenchymal cells by GSTs into [11C]MPG, which is exported from cells by MRP1 and eliminated across the mouse BBB by OAT3 and MRP4 [15]. As opposed to the mouse brain (kE: 1.5 ± 0.06 h− 1) [10, 12], [11C]BMP-derived radioactivity was very slowly eliminated from the human brain, pointing to substantial species differences in the expression of OAT3, MRP4 and possibly other transporters at the mouse and human BBB [16]. This is supported by quantitative proteomics data, which revealed that OAT3 expression was below the limit of detection in human brain capillary endothelial cells [33]. kE values were 50–90 times lower in the human than in the mouse brain and neither of the two analysed brain regions (cortex and cerebellum) showed significant kE changes following probenecid administration (Fig. 5). This contrasts with mouse data, which show a significant kE decrease in the brain after administration of the non-subtype-specific MRP inhibitor MK571 [12, 13]. While it is not certain whether probenecid crossed the BBB in sufficient amounts to inhibit MRP1 in brain parenchymal cells at the administered dose, the slow brain washout of radioactivity suggests that [11C]BMP may not be suitable to measure MRP1 function in the human brain.
Surprisingly, we observed a distinct regional radioactivity uptake pattern in the brain with preferential grey matter accumulation and higher accumulation in cerebellar as compared with cortical grey matter (Fig. 2b). This cerebral uptake pattern closely resembles that of the SPECT tracer [99mTc]Tc-meso-hexamethyl propyleneamine oxime (HMPAO), which is a stereoisomer of the clinically used perfusion imaging tracer [99mTc]Tc-d, l-HMPAO [34]. It has been proposed that [99mTc]Tc-meso-HMPAO crosses the BBB by passive diffusion and is inside the brain converted into a hydrophilic glutathione-conjugate which is retained in brain tissue. As the rate-limiting step in the brain kinetics of [99mTc]Tc-meso-HMPAO is its conversion into the glutathione-conjugate, its regional brain uptake depends on regional glutathione content. Experiments in mice revealed preferential grey matter accumulation of [99mTc]Tc-meso-HMPAO-derived radioactivity, higher accumulation in the cerebellum than in the cortex, and a good correlation between regional brain uptake and regional glutathione concentration [34]. Given that [11C]BMP also undergoes glutathione-conjugation and is trapped in the human brain, it is tempting to speculate that the distinct regional uptake pattern of [11C]BMP-derived radioactivity may be related to regional differences in the glutathione-conjugation system (i.e., glutathione concentration and/or GST activity). Since glutathione content is an important marker of oxidative stress and since no PET tracer for mapping the glutathione-conjugation system in the brain is currently available, further investigations including mechanistic preclinical studies are warranted to assess the suitability of [11C]BMP for this application.
Apart from brain parenchymal cells, MRP1 is expressed in the basolateral (blood-facing) membrane of choroid plexus epithelial cells (Supplementary Fig. 4b), where it restricts the distribution of its substrates from blood into cerebrospinal fluid [35]. Choroid plexus epithelial cells possess high GST activity, which forms together with MRP1 an efficient cellular detoxification system [36]. As opposed to the slow brain kinetics of radioactivity, the choroid plexus showed appreciable washout of radioactivity suggesting the presence of some efflux mechanism (Fig. 4). Indeed, radioactivity concentrations in the ventricular system were 3–4 times lower than in brain tissue, which suggested restricted distribution of radioactivity across the blood-cerebrospinal fluid barrier. However, similar to the brain, probenecid administration had no effect on kE in the choroid plexus (Fig. 5). Since blood capillaries of the choroid plexus are fenestrated, it appears unlikely that insufficient probenecid exposure was the cause for the lack of an effect of probenecid on the kinetics of [11C]BMP-derived radioactivity in the choroid plexus. In contrast, we observed a pronounced kE reduction in the retina (Fig. 5), in which MRP1 is expressed in the plasma membrane of the retinal pigment epithelium forming the outer blood-retina barrier (Supplementary Fig. 4c) [37]. Even though we were not able to correct the retinal TACs for vascular radioactivity content, it appears unlikely that the effect seen in the retina was only caused by a blood effect, as the kE reduction in the retina (− 57 ± 29%) largely exceeded the kE reduction in blood (− 26 ± 10%). The exact reasons for the discrepancy between the effect of probenecid on radioactivity kinetics in the choroid plexus and retina remain unclear.
Even though BMP is not a clinically used drug, it can be considered as a drug-like molecule which has similar physicochemical properties as many small-molecule drugs. Transporter-mediated DDIs are of great concern in drug development and regulatory authorities currently mandate to assess the DDI risk for new drug candidates [3]. When a DDI risk cannot be excluded based on available in vitro data, clinical studies are required. In most clinical DDI studies, drug concentrations are only assessed in plasma and changes in tissue pharmacokinetics, which could for instance cause organ toxicity (e.g., statin-induced rhabdomyolysis), may therefore remain undetected [5]. PET employing microdoses of radiolabelled drugs can be used to safely assess DDIs in humans at a tissue level [5]. Up to now, this has only been possible in single organs due to the limited axial FOV of previously available clinical PET scanners [38]. The availability of LAFOV PET scanners can thus be considered as a major step forward, as it will allow for assessing drug disposition and DDIs in humans at a whole-body, multi-tissue level. In this context, unique advantages of LAFOV PET scanners include the possibility to obtain an image-derived blood input function from a large blood vessel within the FOV of the scanner (i.e., the aorta) and their high sensitivity which potentially allows for longer dynamic scan durations than with conventional PET scanners, thus enabling improved pharmacokinetic analysis.





Comments (0)