Cell Culture
Two PA cell lines, GH1 and RC-4B/C, were purchased from Jennio Biotech (Guangzhou, China). The cells were cultured in RPMI1640 medium supplemented with 100 U/mL penicillin, 100 mg/mL streptomycin, and 10% exosome-free fetal bovine serum (FBS). The exosome-free FBS was produced by centrifugation (100,000 g) at 4 °C overnight to ensure the removal of any bovine-derived exosomes [7]. All cell cultures were incubated under the following conditions: 95% humidity, 5% CO2, and 37 °C.
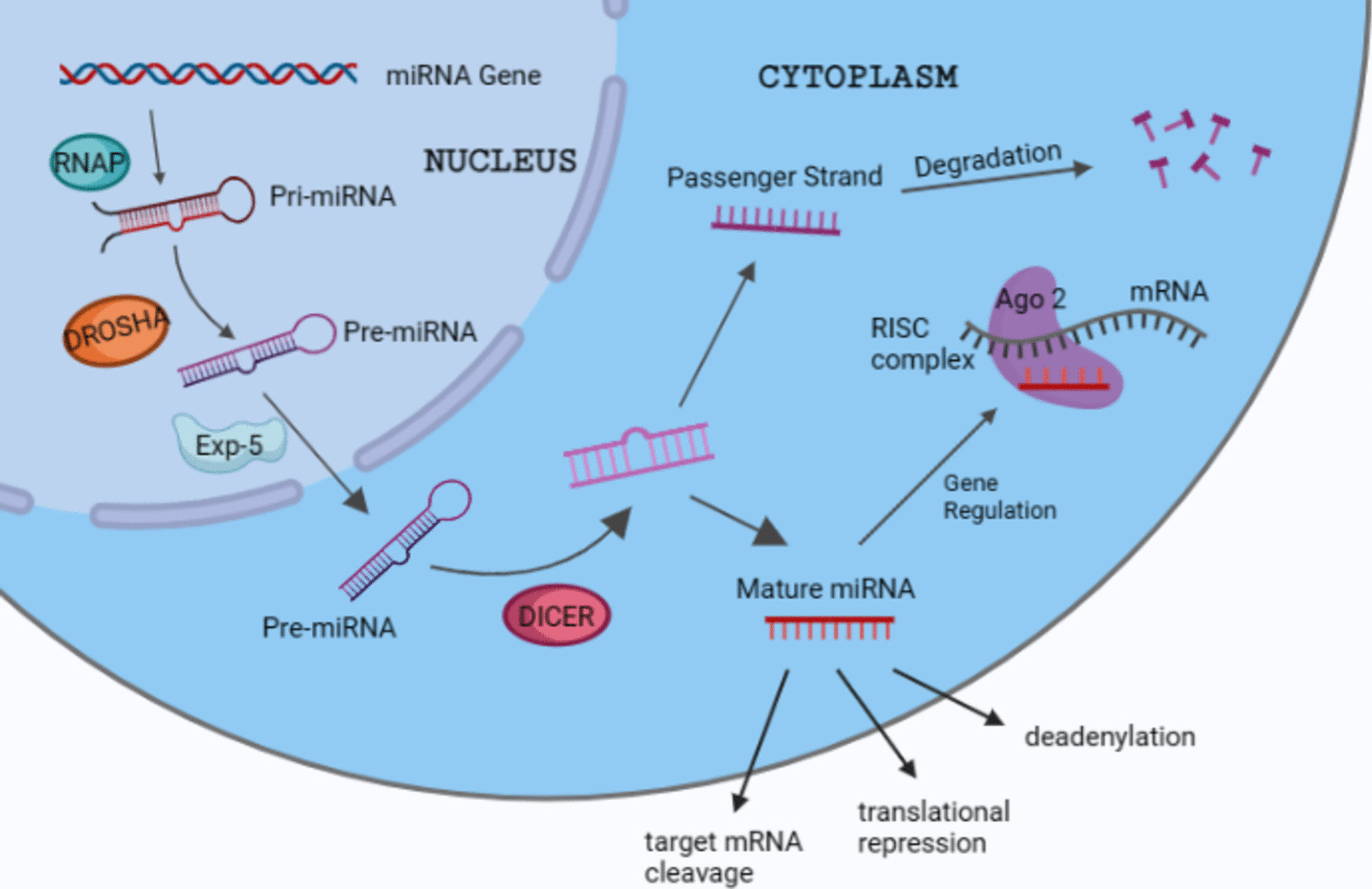
Exosome Isolation
Exosomes were extracted through serial ultracentrifugation procedures. After removing cells and other debris by centrifugation at 300 g and 3000 g at 4 °C, respectively. Fifty milliliters of the supernatants of GH1 and RC-4B/C cells were centrifuged at 10,000 × g at 4 °C for 20 min to remove shedding vesicles and the other vesicles with larger sizes. The resulting supernatant was transferred to a sterile centrifuge tube and then ultracentrifuged at 110,000 × g for 70 min to concentrate exosomes, and exosomes obtained were purified through centrifugation at 110,000 × g for 1 h. The exosomes were then resuspended in phosphate-buffered saline (PBS), filtered through 0.22-mm filters (C8848, Millipore, Boston, MA), and stored at − 80 °C in PBS until further experiments. For each assay, 10 µg of exosomes resuspended in 100 µL 1 × PBS were added to 1 × 105 cells for 24 h.
Transmission Electron Microscopy (TEM)
The purified exosomes were fixed in 2% paraformaldehyde, and a drop of exosome fractions was floated on a nickel TEM grid lined with a formvar/carbon film and a mesh size of 400. The sample was stained with 2% uranyl acetate and examined on a TEM (Hitachi H7600, Japan) at 80 kV.
Nanoparticle Tracking Analysis (NTA)
The sizes and numbers of exosomes were determined on the NanoSight NS300 system (Malvern Instruments, Westborough, MA, USA). The exosomes obtained were suspended in PBS and then diluted 100–500 times to achieve a concentration of 20 to 100 objects per frame. Subsequently, they were injected into sample chambers at room temperature, which were equipped with a 488-nm laser and a high-sensitivity sCMOS camera. Each video was analyzed by examining a minimum of 200 completed tracks, and the data were processed using NTA analytical 2.3 software (Shanghai XP Biomed Ltd., Shanghai, China).
Exosome Concentration Measurement
ExoELISA Ultra CD81 assay (System Biosciences) was used to measure indirectly exosome concentration. Total protein was extracted from exosomes using the RIPA buffer with protease inhibitor (Thermo Fischer Scientific, Waltham, MA, USA) and quantified using the BCA kit. A total of 500 µg of total protein of each sample and CD81 standards was loaded onto a 96-well plate and incubated for 1 h at 37 °C. The wells were incubated with an anti-CD81 primary antibody at room temperature (RT) for 2 h and then incubated with a horseradish peroxidase-conjugated secondary antibody at RT for 1 h. Fifty microliters of tetramethylbenzidine substrate was added to each well and incubated for 10 min at RT. Stop buffer was added and absorbance was measured at 450 nm using a microplate reader (Thermo Fischer Scientific) [18].
Cell Transfection
Short hairpin RNA (shRNA) targeting AFAP1-AS1 (sh-AFAP1-AS1-1: CCGGGAGTACATCACCTCAAATTATCTCGAGATAATTTGAGGTGATGTACTC-TTTTTT; sh-AFAP1-AS1-2: CCGGAGGCAGAACTGGAGAAGAAATCTCGAGATTTCTTCTCCAGTTCTGCCTTTTTTT), and negative control (NC: CCGGTTCTCCGAACGTGTCACGTCTCGAGACGTGACACGTTCGGAGAATTTTTT) lentivirus were purchased from Genechem (Shanghai, China) and were used to infect PA cell lines in the presence of polybrene (Sigma-Aldrich, St. Louis, MO, USA) at a concentration of 8 µg/mL. Puromycin (Thermo Fisher Scientific, Waltham, MA, USA) was included during transfections to select stable clones. Transfection efficiency was verified by quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR). Wild-type AFAP1-AS1 and mutant plasmids, as well as overexpressing HuR, were constructed by Genechem. Small interfering RNA SMURF1 (si-SMURF1) and si-NC were obtained from Genechem. PA cells were transfected using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions. The sequences of oligonucleotides and plasmids were listed in Supplementary Table 1 and Materials and methods.
Cell Counting Kit-8 (CCK-8) Assay
Briefly, about 3000 cells were seeded into 96-well plates and allowed to incubate overnight. They were treated with various agents as follows: control exosomes, AFAP1-AS1-knockdown exosomes, HuR plasmid, or a combination of AFAP1-AS1-knockdown exosomes and HuR plasmid, for 0, 24, 48, and 72 h. CCK-8 reagent (Dojindo, Kumamoto, Japan; 10 µL/well) was added to the cells at specified time points and incubated for 2 h, after which the absorbance was read at 450 nm using a microplate reader (Thermo Fischer Scientific).
Colony Formation Assay
Cells were seeded in 6-well plates, at a density of 1000 cells per well and incubated for 2 weeks at 37 °C. Next, the cells were fixed with 4% paraformaldehyde and subsequently stained with 0.5% crystal violet.
Wound Healing Assay
PA cells were first seeded in 6-well plates and incubated at 37 °C until they reached 100% confluency. Subsequently, a pipette tip was used to create a scratch wound, and wound closure was imaged after 48 h using a microscope equipped with a digital camera. Relative migration distance was measured using ImageJ software, by determining the fraction of cell coverage across the scratch and calculated using the following formula: (%) = migration area/total area × 100%.
Transwell Assay
Transwell assays were conducted using matrigel-coated invasion and polycarbonate membrane inserts with 8-µm pore size. Approximately 1 × 104 cells were added to the upper chamber, while 10% FBS was administered to the lower chamber. After 24 h, the chambers were fixed and stained with 0.05% crystal violet for 2 h. The cells in the upper surface were carefully scraped off, stained, and observed under a microscope equipped with a digital camera.
RNA Extraction and qRT-PCR
Total RNA was extracted from PA cell lines using the TRIzol reagent following the manufacturer’s instructions. The RNA was reverse transcribed to cDNA using the PrimeScript RT reagent Kit (Takara, China) and subjected to qRT-PCR using the SYBR-Green PCR Master Mix (Takara, China). Amplification was performed on the applied Biosystems Quant Studio system (Thermo Fisher Scientific). Expression of the target genes was analyzed using the 2−ΔΔct method and normalized to that of β-actin. Primer sequences are listed in Supplementary Table 1.
Northern Blotting
Ten micrograms of total RNA was separated by electrophoresis and then transferred to nitrocellulose filter membranes. Membranes were incubated with the hydration buffer containing biotin-labeled AFAP1-AS1 or 18S rRNA probes at 55 °C overnight. Finally, the RNA signal was detected using the Chemiluminescent Nucleic Acid Detection Module (Thermo Fisher Scientific). The sequences of biotin-labeled probes were listed in Supplementary Materials and methods.
Subcellular Fractionation
Nuclear and cytoplasmic RNA was isolated using the nuclear or cytoplasmic RNA purification kit (Thermo Fischer Scientific), following the manufacturer’s instructions. The cells were first lysed on ice for 10 min using cell fraction buffer and then centrifuged for 5 min at 500 g to separate cytoplasmic and nuclear fractions. The extracted RNA was converted to cDNA and subjected to qRT-PCR, utilizing β-actin and U6 as markers for cytoplasmic and nuclear compartments, respectively.
RNA-Fluorescence In Situ Hybridization (FISH)
This was performed using the Fluorescent In Situ Hybridization Kit (Ribo, Guangzhou, China), according to the manufacturer’s instructions. Briefly, cells were seeded onto cover slides, and AFAP1-AS1 detection was achieved using a specific probe labeled with Cy3. Detection was done on a Nikon Eclipse Ti microscope equipped with a digital camera (Nikon, Kanagawa, Japan).
Extracellular Acidification Rate (ECAR) and Mitochondrial Oxygen Consumption Rate (OCR) Assays
ECAR and OCR assays were performed using the Seahorse XF96 Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA, USA) following the manufacturer’s instructions. Briefly, 8000 cells/well were seeded in a Seahorse XF96 culture microplate, rinsed with Seahorse buffer, and then mixed with Seahorse buffer supplemented with oligomycin (1 µM), FCCP (1 µM), or rotenone (1 µM) to measure OCR. Similarly, glucose (10 mM), oligomycin (1 µM), or 2-deoxy-glucose (2-DG, 100 mM) was added to measure the ECAR. The obtained data were analyzed using the Seahorse software. Cells were digested with trypsin and counted with a hemocytometer. ECAR and OCR were normalized to cell numbers.
Glucose Uptake
The uptakes of glucose were measured using the fluorescent 2-DG analog 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino)-2-deoxyglucose (2-NBDG). The cells were stained with 2-NBDG (10 µM) for 1 h, and washed twice by PBS. Luminescence intensity was read by a microplate reader (Thermo Fischer Scientific, Waltham, MA, USA). Cells were digested with trypsin and counted with a hemocytometer. Glucose uptake was normalized to cell numbers.
Lactate Assay
Lactate concentration in cell supernatants was measured using a lactate assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China), according to the manufacturer’s instructions. Cells were digested with trypsin and counted with a hemocytometer. Lactate concentration was normalized to cell numbers.
Mice Xenograft Assay
Thirty male NOD-SCID mice (6 weeks) were procured from the Laboratory Animal Center of Southern Medical University (Guangzhou, China) and randomly allocated into five groups (6 mice/group) prior to inoculation. Control (cells were cultured in RPMI1640 medium containing exosome-free FBS), exosomes, sh-NC exosomes, sh-AFAP1-AS1-1 exosomes, and sh-AFAP1-AS1-2 exosome–treated GH1 luciferase cells (1 × 106) in 0.2 mL PBS were delivered into the tail veins of NOD-SCID mice. Cell metastasis was monitored weekly through bioluminescence imaging on the IVIS system (Xenogen, Alameda, CA, USA), with mice receiving 150 mg/kg d-luciferin potassium salt (Caliper Life Sciences, Hopkinton, MA). After 4 weeks, the representative bioluminescence imaging of metastases was measured, and the lung tissues were collected for hematoxylin–eosin staining (H&E staining) [7]. The protocol used in animal experimental procedures was approved by the Institutional Animal Care and Use Committee of Guangzhou First People’s Hospital, South China University of Technology (K-2021–041-01).
H&E Staining
Paraffin-embedded lung tissue sections from mice were dewaxed via heating at 65 °C for 2 h, hydrated, and then cell nuclei and cytoplasm stained with hematoxylin and eosin solution (Biosharp, Anhui, China), respectively. The slices were then dried and preserved in neutral resin (SCR, Shanghai, China).
Cycloheximide (CHX) Chase Assay
AFAP1-AS1 knockdown exosome–treated GH1 cells were treated with cycloheximide (50 mg/mL) from 0 to 12 h, proteins extracted from the cell lysates using a lysis buffer containing 2% sodium dodecyl sulfate (SDS), separated via SDS–polyacrylamide gel electrophoresis (PAGE), followed by western blot targeting the HuR antibody.
Western Blot and Coimmunoprecipitation (co-IP) Assays
Proteins were extracted from PA cell lines using the RIPA buffer with protease inhibitor (Thermo Fisher Scientific) and quantified using the BCA kit. The proteins were separated on a 10% SDS-PAGE and transferred to polyvinylidene fluoride membranes (Millipore, USA). The membranes were hybridized overnight with primary antibodies CD9 (1:1000; Abcam, Cambridge, MA, USA), CD81 (1:1000; Proteintech, China), TSG101 (1:1000; Abcam), Alix (1:1000; Affinity, China), CDK2 (1:1000; Abcam), Cyclin D1 (1:5000; Proteintech), p21 (1:1000; Abcam), p27 (1:1000; Affinity), N-cadherin (1:5000; Abcam), Vimentin (1:1000; Abcam), MMP9 (1:1000; Abcam), E-cadherin (1:1000; Abcam), HuR (1:1000; Abcam), SMURF1 (1:1000; Proteintech), hexokinase 2 (HK2) (1:1000; Abcam), calnexin (1:1000; Abcam), and pyruvate kinase M2 (PKM2) (1:1000; Abcam) at 4 °C and then incubated with a secondary antibody at RT for 1 h. Finally, the protein blots were detected utilizing an ECL kit. For the co-immunoprecipitation (co-IP) assay, proteins were extracted using the Co-IP lysis buffer (Bersinbio, Guangzhou, China, Bes3011) with protease inhibitor (Thermo Fisher Scientific) and quantified using the BCA kit. A total of 500 µg of proteins were incubated overnight with primary antibodies against HuR (Abcam) and SMURF1 (Proteintech) at 4 °C, followed by a 4-h incubation with protein A/G beads at the same temperature. The immunoprecipitated proteins were subsequently eluted from the beads and subjected to western blot analysis using the specified antibodies.
Ubiquitination Assay
AFAP1-AS1-knockdown exosome–treated cells were treated with MG-132 (20 µM) for 8 h, proteins extracted using the Co-IP lysis buffer (Bersinbio, Bes3011), and incubated for 3 h with anti-HuR antibody at 4 °C. The proteins were incubated overnight with A/G beads (Invitrogen) at 4 °C. Precipitate protein complexes were subjected to western blot with an anti-ubiquitin antibody (Cell Signaling Technology, Beverly, MA, USA).
RNA Pulldown Assay
Cells were transfected for 24 h with biotin-labeled AFAP1-AS1 (GenePharma, Shanghai, China), lysed using lysis buffer (Bersinbio, Bes5102) containing magnetic beads, and incubated at room temperature with gentle rotation for 30 min. Next, the biotin-coupled RNA–coated beads were washed 4 times, purified, and subjected to western blot assay. The sequences of biotin-labeled AFAP1-AS1 probes were listed in Supplementary Materials and methods.
RNA-Binding Protein Immunoprecipitation (RIP) Assay
Cells were lysed with RIP lysis buffer, using magnetic beads washed with RIP wash buffer (Bersinbio, Bes5101), followed by the addition of anti-HuR (Abcam) and anti-IgG antibodies, and then incubated for 30 min at room temperature. Total RNA was used as a control during the RIP process. The samples were incubated overnight at 4 °C and washed 5 times with the RIP wash buffer. Total RNA was extracted using the TRIzol method, reverse transcribed to cDNA, and then subjected to qRT-PCR to determine the expression of AFAP1-AS1.
Statistical Analysis
Data were statistically analyzed using SPSS 22.0 (IBM, Chicago, USA) and GraphPad Prism 9.0 (San Diego, USA) software and presented as means ± standard deviation. Comparisons between and among groups were performed using Student’s t-test and analysis of variance, respectively, and data with P-values of less than 0.05 were considered statistically significant.







Comments (0)