Animals and ethical approval
Eight-week-old male C57BL/6J mice were purchased from Shanghai Model Organisms Center, Inc. in Shanghai, China. The Shanghai Model Organisms Center, Inc. (SMOC) IACUC approved all animal experimental plans (July 20, 2024, Approved Number: 2024–0014). ARVO's Statement for the Use of Animals in Ophthalmic and Visual Research was strictly followed for every experimental methodology and animal husbandry protocol. Before conducting the experiments, the animals were allowed to adapt to the housing conditions spanning a duration of 7 days or more.
Laser induced CNV model
Mice were anesthetized with 2% pentobarbital sodium solution administered intraperitoneally at 80 mg/kg (provided by SMOC, China), and the pupils were dilated using Compound Tropicamide Eye Drops. After fixing the eye in the appropriate position, a 5.4-mm handheld contact lens was placed over the cornea. A 532-nm frequency-doubled argon laser (parameters: power 100 mW, spot diameter 50 μm, exposure time of 100 ms) was then used to photocoagulation burns created circumferentially around the optic disc, 1–1.5 PD away from its margin. Following photocoagulation, a bubble (devoid of retinal hemorrhage) is formed, leading to disruption of the Bruch membrane, indicating effective establishment of a coagulated point.
Vitreous delivery via injection
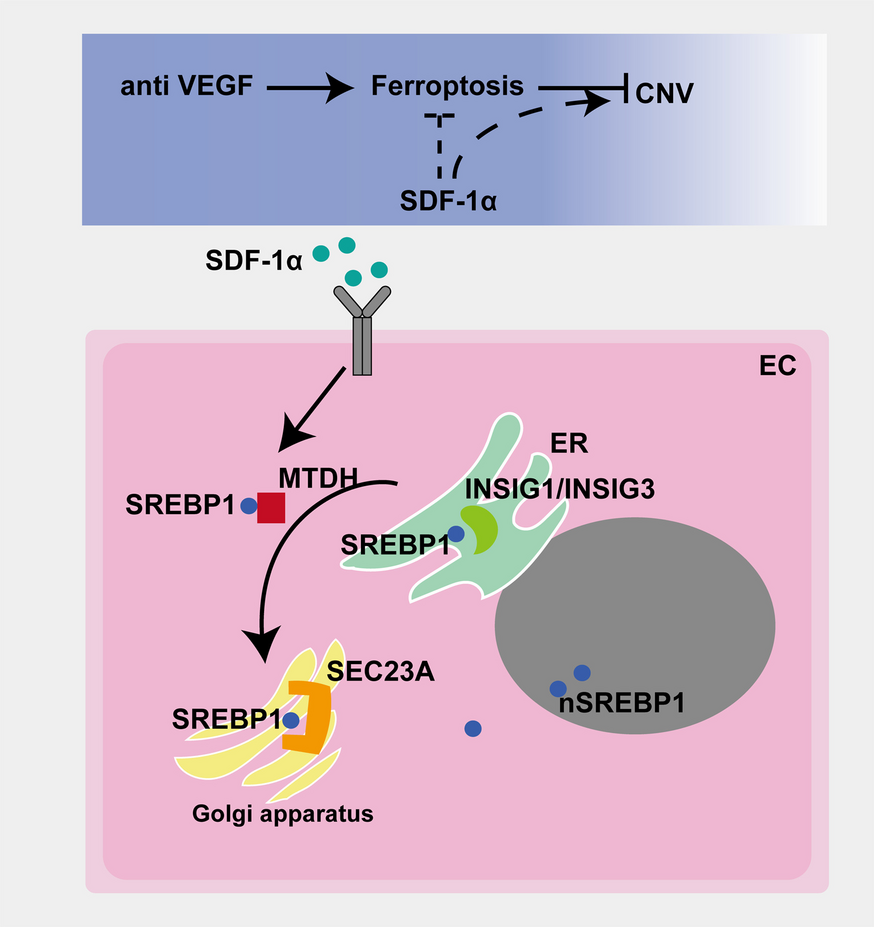
Under anesthesia, the mice were given eye antibiotic eye drops, and a 33-gauge syringe was vertically inserted to administer Anti-VEGF agent Ranibizumab (Novartis Pharma Schweiz AG) (1μL), Erastin (Selleck, USA, #S7242) (1μL) and SDF-1α (Biolegend, USA, # 589,806) (1uL) into the vitreous cavity via the equator of the eyeball.
Retinal flat-mounting and immunofluorescence staining
Eyeball samples from mice were harvested and immersed in 4% Paraformaldehyde fixative for 3 h. With the help of an optical microscope, the retinas were carefully isolated. Then, for permeabilization and blocking, the retinas were subjected to overnight incubation at 4 °C in a solution containing 1% BSA and 0.5% Triton X-100. Next, the retinas were soaked in a solution containing FITC-labeled Lectin B4 (dilution ratio 1:100) (Vector labs, USA, # DL-1207) for an overnight duration at 4 degrees Celsius. PBS was utilized for three separate washing cycles and retinas were trimmed into a four-leaf clover shape. Then, the retinas were sealed with a fluorescent-protecting agent and stored at 4 ℃ in an environment free of ambient light. Finally, the retinas were observed, and photos were taken, using a laser confocal microscope.
Tissue immunofluorescence staining
Histologically sections were taken of the eye and optic nerve. After dewaxing and antigen retrieval, a 5% BSA blocking buffer was applied to the tissues for 60 min. Thereafter, the tissues were probed with primary antibodies and incubated for 12 h (diluted as per the manufacturer’s recommended procedures) against the following proteins at 4 °C overnight: SREBP1(1:100, Invitrogen, USA, # PA1-337), GPX4 (1:100, Proteintech, China, #67,763–1-Ig), xCT (1:100, Proteintech, China, #26,864–1-AP), TFRC (1:100, Abcam, UK, #ab214039). Following three washes with PBS (5 min per wash), were then incubated with species-matched fluorescent secondary antibody. Then, the tissues were sealed with fluorescence-protecting agent and stored at 4 ℃ in an environment free of ambient light. The tissues were observed, and photos were taken, using a laser confocal microscope (Olympus FV3000).
Cell and cell culture
The bEND.3 mouse brain microvascular endothelial cell line (BMECs), derived from murine brain tissue, was acquired from ATCC (CRL-2299). Cells between passage 11 and passage 30 are used for experiments.
Ranibizumab treatment: For subsequent experiments, BMECs were plated into 6-well plates at a seeding density of 3.6 × 105 cells per well. In a standard culture setup, in a humidified 5% CO₂ incubator at 37 °C, BMECs were cultured in DMEM medium consisting of 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin for 24 h. On the second day, the growth medium was substituted with low-serum DMEM (2% FBS) for cell synchronization and Ranibizumab (1 μL/mL) for 24 h for treatment. For the blank control group, only complete medium (without Ranibizumab) is added. Other operations (such as cell density and culture conditions) are exactly the same as those in the Ranibizumab treatment group.
Erastin, RSL3, SDF-1α treatment: 3.6 × 105 cells per well of BMECs were planted into a 6-well plate and 3.0 × 104 cells per well of BMECs were planted into a 24-well plate. Cell culture conditions followed the earlier protocol in Ranibizumab treatment. On the second day, DMEM containing 2% FBS and Erastin (10 mM in DMSO) (Selleck, USA, #S7242), RSL3 (1 Mm in DMSO) (MedChemExpress, USA, # HY-100218A), or SDF-1α (100 μmol/L in distilled water) (Biolegend, USA, # 589,806) was used to renewed the culture medium, followed by a 24-h culturing period. For the groups treated with Erastin and RSL3, a volume of DMSO equal to that in the experimental group was pipetted into the control group’s medium. For the group treated with SDF-1α, the same volume of sterile distilled water was incorporated into the culture medium as part of the control group’s treatment protocol.
After 24 h, the cells were harvested, and RNA and protein isolation was performed for subsequent qRT PCR, immunoblotting detection or cellular immunofluorescence staining.
siRNA transfection: 3.6 × 105 cells per well of BMECs were seeded into six-well plates. Then, 20 μM siRNA (siVEGFR2, siSREBP1, siMTDH) was added to OPTI-MEM Reduced Serum Medium (Gibco, USA) and mixed with Lipofectamine™ regent (ThermoFisher Scientific, USA, # L3000-008), and the mixture was added to the growth medium in the wells. After 48 h, proteins were collected and cell functions were assessed. The sequences of siRNA of VEGFR2, SREBP1 and MTDH used in the experiment are shown in Tables 1, 2 and 3:
Table 1 siRNA sequences targeting VEGFR2Table 2 siRNA sequences targeting SREBP1Table 3 siRNA sequences targeting MTDHRNA isolation and quantitative PCR
Cellular RNA was isolated using the RNA Extraction Kit (KeyGEN BioTECH, #KGR203) in strict accordance with the kit’s protocol. Subsequently, PCR amplification was carried out with TB Green® Premix Ex Taq™ (Takara Bio, Japan, #RR820A) on a fluorescence-based qPCR system. We programmed the thermal cycler with an initial 95 °C denaturation for 30 s, followed by 40 amplification cycles comprising 3 s at 95 °C and 30 s at 60 °C for annealing/extension. β-actin served as the housekeeping gene for normalization. To analyze the gene expression levels, we normalized the Ct values of each target gene to the corresponding β-actin Ct values from the same sample. Then, fold changes in gene expression were derived by calculating 2 − ΔΔCt, where ΔΔCt = (CtTarget − CtReference) Treated − (CtTarget − CtReference) Control. The results were presented as relative fold changes, with the control group set as the baseline. All primer sequences used in this study are listed in Table 4.
Table 4 Primer sequences used in RT-qPCRImmunoblotting
Proteins were collected from endothelial cells, and retina -choroid-sclera complex by Radioimmunoprecipitation Assay Lysis Buffer (RIPA) (KeyGEN BioTECH, Nanjing, China, #KGB5203-100) containing proteolytic enzyme inhibitors (Epizyme Biotech, Shanghai, China, #GRF101) and phosphatase-blocking agents (Epizyme Biotech, Shanghai, China, #GRF102). We calculated the loading volume for 40 µg of total protein using a BCA assay kit (KeyGEN BioTECH, Nanjing, China, # KGB2101-500). Protein samples were subjected to electrophoresis on a 12% SDS-PAGE gel (Epizyme Biotech, Shanghai, China, #PG113) using Tris–glycine running buffer (Epizyme Biotech, Shanghai, China, #PS105) at a constant voltage of 80 V for the first 30 min, then 120 V for the subsequent 60 min. Subsequently, proteins were transferred onto a PVDF membrane via wet transfer at a constant current of 200 mA for 2 h in the transfer buffer (Epizyme Biotech, Shanghai, China, #PS109). Then, the PVDF membrane (Millipore, Massachusetts, USA, #ISEQ00010) was immersed in a blocking solution containing 10% non-fat dried milk, reconstituted in Tris-buffered saline containing Tween 20 (TBST) (Epizyme Biotech, Shanghai, China, #PS103)) and gently shaken for 1 h at ambient temperature. Target-specific antibodies were judiciously selected in strict accordance with the defined experimental goals and pre-defined detection metrics stipulated in the research protocol (showed in Table 5). Working solution were used to incubate the PVDF membrane at 4 ℃ overnight. After washing the PVDF membrane with TBST solution, Horseradish peroxidase (HRP) labeled secondary antibodies incubate membrane at ambient temperature for a duration of 1 h. Washed the PVDF membrane with TBST solution again. Finally, the PVDF membrane was immersed in ECL reagent (Epizyme Biotech, Shanghai, China, #SQ201L) and incubated in the dark for 1 min. The chemiluminescence of protein bands in the PVDF membrane was observed using a Tanon 4600 series automatic imaging system for chemiluminescence and fluorescence analysis, and photographs were taken. Subsequently, Image J software was used to quantify the gray values.
Table 5 The antibodies used and their dilution ratiosImmunoprecipitation
The endothelial cells collected from the treatment and control groups in 1.5 mL centrifuge tubes underwent lysis through immunoprecipitation lysis buffer (Epizyme Biotech, Shanghai, China, #PC105) containing cocktail of inhibitors of protease (Epizyme Biotech, Shanghai, China, #GRF101) was added at a ratio of 30 μL per 1.0 × 105 cells. After mixing, ice incubation of the samples was conducted for 20 min. Subsequently, the protein-containing lysate was collected by centrifugation at 12,000 × g for 10 min at 4 °C. The antibody (SREBP1 (Santa cruz, USA, #sc-365513), MTDH (Abcam, UK, #ab227981)) and protein A/G magnetic beads (Epizyme Biotech, Shanghai, China, #YG003) were sequentially added to the cell lysis buffer. The EP tubes were mounted on a rotator and subjected to incubation at room temperature for 2 h. Then clear, bead-free liquid was decanted and discarded and bead washing was carried out by immunoprecipitation lysis buffer. Cleaned magnetic beads were resuspend using immunoprecipitation lysis buffer containing protease inhibitors and the protein was diluted with loading buffer and then denatured by heating at 100 °C to facilitate subsequent Western blot detection.
Cell proliferation assay
Cell viability was measured by CCK8 detection kit (KeyGEN BioTECH, Nanjing, China, # KGA9306-1000). Cells in each well of the 96 well plate were added with 10μL of CCK8 staining solution and placed in 37 ℃, 5% CO2 cell culture incubator for about 90 min. Lastly, we determined the OD value at 450 nm using the SpectraMax® iD3 continuous spectral multi-mode microplate detection platform from Molecular Devices. Calculate the proliferation activity of each group of cells using the formula: (Experimental well – Blank well)/(Control well – Blank well) × 100%.
Cell migration assay using wound-healing method
When the cells had reached full confluency in six-well plates, straight lines were drawn in confluent cell monolayer along a ruler. After three washes using PBS, the cells were cultured in serum-reduced medium containing 1% FBS to minimize serum-induced effects. After 0, 1, 2, 3, 4, 6, 12, and 24 h, the cells were observed, and photos were taken, using an OLYMPUS BX63 microscope. Image J software was used to measure the scratch width and perform quantitative analysis.
Matrigel tube formation assay
The Matrix-Gel™ Basement Membrane Matrix (Standard, Phenol Red-free) (Beyotime, Shanghai, China, # C0372) was removed from a −20 °C refrigerator to a 4 °C refrigerator until it thaws and melts into a liquid. Endothelial cells of control group and treated group were isolated and collected by 0.25% Trypsin–EDTA Solution (Phenol Red-free) (KeyGEN BioTECH, Nanjing, China, # KGL2101-100). Resuspend cells in complete culture medium and made sure cell density was 6 × 104 cells/ml. Add 100 μL of Matrix-Gel™ Basement Membrane Matrix to the bottom of a 96-well plate, avoiding bubble formation. Then incubate the plate in a 37 ℃ incubator for 30 min to make the Matrix solid. 100 μL of cell resuspension was added to the surface of the Matrigel for 4 h’ incubation. Images were acquired using an OLYMPUS BX63 microscope. Image J software was utilized to determine the branches.
ER and Golgi fluorescent probe incubation and cellular immunofluorescence staining
ER-Tracker Red probe (Beyotime, Shanghai, China, # C1041) and Golgi-Tracker Red probe (Beyotime, Shanghai, China, # C1043) were utilized in compliance with the kit’s instructions. First, we prepared the probe ready-to-use solution. Then, the medium was replaced with an aliquot of the working solution. After incubation, the cells were briefly fixed with a 4% paraformaldehyde solution. Then, the cells were blocked for 1 h, immersed in 5% BSA. Different antibodies targeting the target protein were added to cells (selected purposely chosen based on the pre-defined experimental goals and detection markers outlined in the research protocol) and placed at 4℃ overnight in the dark. After three PBS washes to remove unbound reagents, the cells were immersed in Alexa Fluor®-conjugated secondary antibodies for signal visualization. Then, the cells were sealed with a fluorescence-protecting agent and stored at 4 ℃ in dark. A laser confocal microscope (Olympus FV3000) was employed to detect images.
Observation changes in mitochondrial morphology in endothelial cells using transmission electron microscopy (TEM)
Digest control and treatment group endothelial cells with 0.25% Trypsin–EDTA Solution (Phenol Red-free) to prepare single-cell suspensions. Collect cells by low-speed centrifugation of the cell suspension, and fix them with pre-cooled 2.5% glutaraldehyde for 2 h to stabilize mitochondrial structures within the cells. Next, rinse the cells 3 times with 0.1 M phosphate buffer for 15 min each to remove excess fixative. Post-fix the cells with 1% osmium tetroxide (OsO₄) for 1–2 h to enhance contrast. Subsequently, dehydrate the cells through a gradient of ethanol solutions (30%, 50%, 70%, 80%, 90%, 100%), with each concentration applied for 10–15 min. After dehydration, replace the ethanol with acetone twice, each for 10 min. Embed the cells in epoxy resin and polymerize them sequentially at 37 °C, 45 °C, and 60 °C. Use an ultramicrotome to cut ultra-thin Sects. (50–70 nm thick), mount the sections onto copper grids, and stain them with uranyl acetate and lead citrate for double staining. Finally, place the grids under a transmission electron microscope, focus stepwise from low to high magnification, observe the ultrastructural features of mitochondria (such as distribution, size, and cristae morphology within cells), and photograph the results for recording.
Analysis of phosphorylation dynamics of proteins in endothelial cells by global phosphoproteomics
Collect cells with good viability, divide them evenly into a control group and an experimental treatment group, with 3 biological replicates for each group to reduce experimental errors and enhance the reliability of the results. After the treatment is completed, collect cell samples from each group separately. We discarded the culture medium and washed the cells three times with pre-cooled PBS. Then, cells were lysed on ice for 15–20 min with lysis buffer containing a protease inhibitor cocktail and phosphatase inhibitors. Meanwhile, perform ultrasonic disruption to further lyse the cells, fully release the intracellular proteins, and prevent changes in phosphorylation modifications. After lysis, centrifuge the samples at 12,000—15,000 rpm at 4 °C for 15—20 min. Collect the supernatant, measure the protein concentration using the BCA method, and adjust the protein concentrations of all samples to the same level.
Next, perform preliminary protein separation by SDS—PAGE electrophoresis. After electrophoresis, divide the gel lanes into several gel pieces according to the molecular weight range (e.g., each interval of 10—20 kDa) to ensure coverage of the entire proteome. Digest the gel pieces with trypsin to obtain a mixture of peptides. Employ TiO₂ (titanium dioxide) enrichment technology to specifically enrich phosphorylated peptides and remove interference from non—phosphorylated peptides. After desalting the enriched phosphorylated peptides, perform liquid chromatography—tandem mass spectrometry (LC—MS/MS) analysis. Process the mass spectrometry data using software such as MaxQuant and Proteome Discoverer. Identify phosphorylation sites by comparing with protein databases, and combine with TMT quantitative technology to analyze the differences in global proteome phosphorylation levels between the control group and the experimental treatment group.
Reactive oxygen species (ROS) production analysis
One day prior to treatment, 50,000 cells were cultured into every well of 24-well culture dishes. Before treatment, cells were firstly immersed in diluted H2DCFDA (25 µM/500 µL Hanks Balanced Salt Solution (HBSS, Gibco®)) (Invitrogen™, USA, Cat. # C6827) at 37 °C for 30 min. Following it, the cells were subjected to corresponding drug or reagent treatments. Lastly, cellular observations and imaging were conducted using an OLYMPUS BX63 microscope.
Assessment of lipid peroxidation by measuring malondialdehyde (MDA)
A lipid Peroxidation MDA Assay Kit (Beyotime, China, # S0131) was employed to determine the level of lipid peroxidation. One day prior to treatment, 200,000 cells were seeded into every well of six-well dishes. Upon completion of the 24-h treatment, the cells were collected using lysis buffer. The thiobarbituric acid (TBA) working solution was prepared, added to the cell lysis buffer, and mixed them. The mixture was heated at 100 °C for 15 min. Then, transfer 200 μL of the reagent into a 96-well plate and measure the OD value at 532 nm using a spectrophotometer.
Statistical analysis
Data were derived from at least three independent experiments and are shown as the mean ± standard deviation (Mean ± SD). For quantitative data comparison between the control and treated groups, the independent samples t-test was employed. To assess data distribution, normality was verified via the Shapiro–Wilk test (P > 0.05), while Levene’s test (P > 0.05) was used to confirm homogeneity of variances. All results are presented as mean ± SD, and statistical analyses were performed using SigmaPlot 14.0 (Systat Software, San Jose, CA). A two-tailed P value < 0.05 was considered statistically significant.







Comments (0)