
Remember me
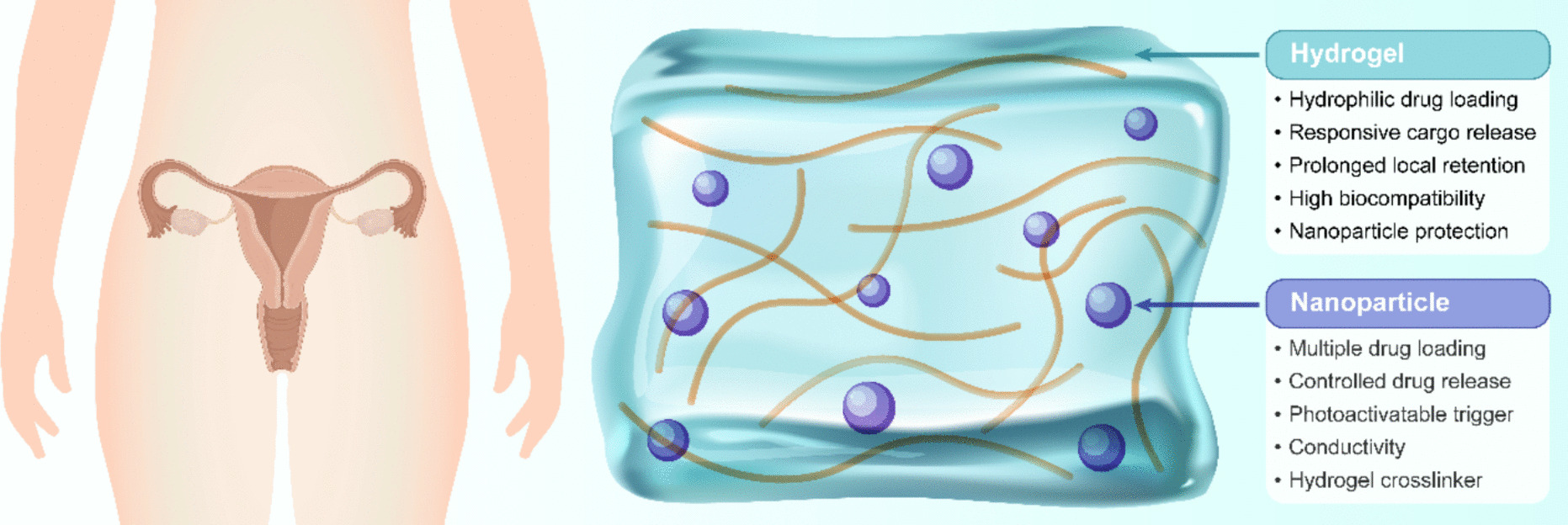






In this study, the freeze-dried FITC-labeled or unlabeled P6 (WYKGDKFLYLQNGIEAIADF) used was provided by Guoping Pharmaceutical Co., Ltd. The P6 stock solution was dissolved in sterile PBS and preserved at −80 °C.
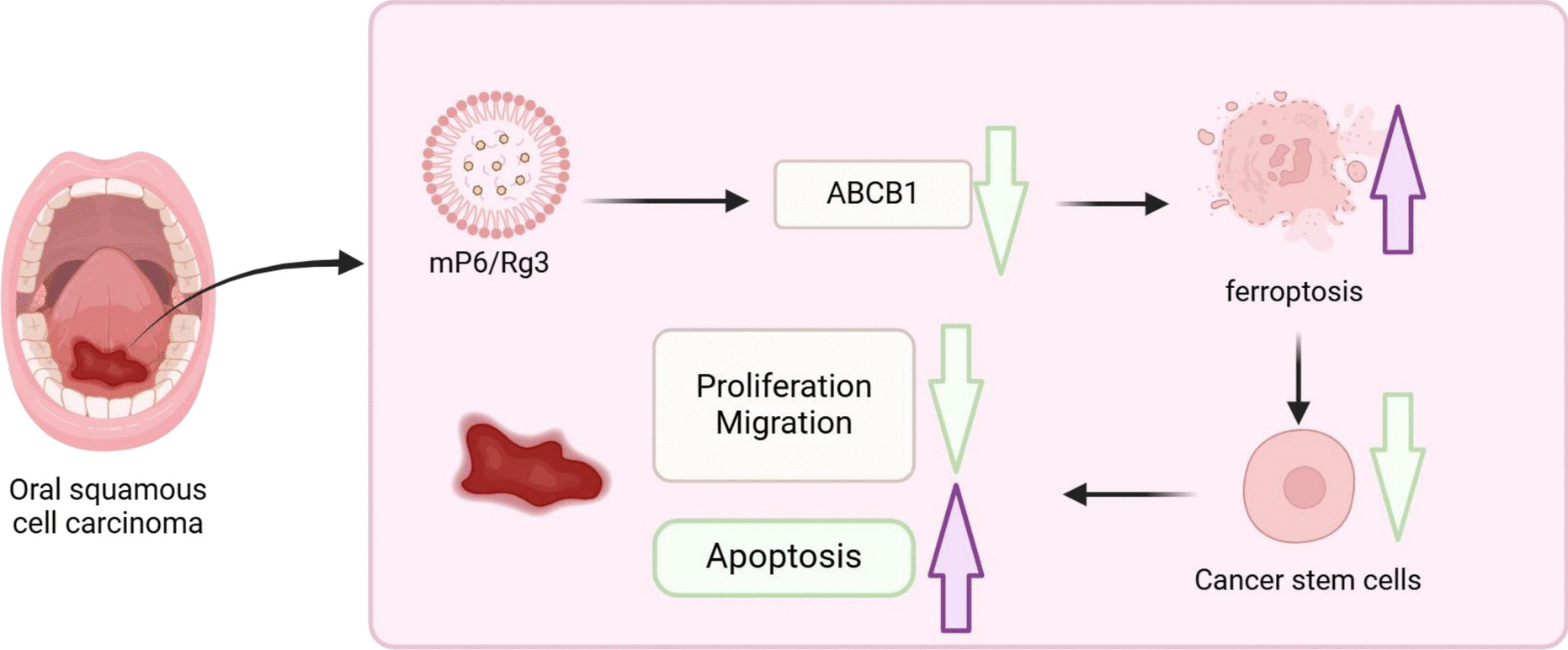
Preparation and characterization of P6-loaded micelles (mP6)/Rg3Initially, FITC-labeled or unlabeled P6 peptides were dissolved in RPMI 1640 medium (11875119, Gibco, USA), while Soluplus polymer (polyethylene caproamide–polyvinyl acetate–polyethylene glycol graft copolymer; M43956, Abmole, USA) was solubilized in sterile PBS. These two solutions were then combined at various mass ratios (ranging from 10:1 to 1:1, Soluplus:P6) using a one-step self-assembly technique to generate mP6 nanomicelles. The resulting micellar solutions were assessed for hydrodynamic diameter and zeta potential in RPMI 1640 medium using a nanoparticle size analyzer (Malvern Instruments Ltd., UK). For free peptide quantification, mP6 samples with a 5:1 polymer-to-peptide ratio were filtered through Amicon ultra-centrifugal units (MWCO 100,000; Millipore) at 12,000 g, 4 °C for 20 min. The filtrate was analyzed for unincorporated P6 content by recording the absorbance at 495 nm with a UV–Vis spectrophotometer (LAMDBA 950, Perkin-Elmer, USA).
The loading of Rg3 was conducted as follows: 200 μL of Rg3 (2 mg; 08511, Sigma Aldrich) solution was added to 1 mL of mP6 suspension (10 mg). The mixture was gently stirred in the dark at ambient temperature for 12 h. Subsequently, the mixture was centrifuged at 1 × 104 rpm for 10 min to eliminate unbound Rg3, and the sediment was redispersed in 2 mL of deionized water to obtain Rg3-loaded nanomicelles, denoted as mP6/Rg3. Unencapsulated Rg3 in the supernatant was examined at 425 nm utilizing a UV spectrophotometer (UV-1900, Shimadzu). Drug loading capacity (DLC) and drug loading efficiency (DLE) were determined as follows: DLC (%) = (drug mass in micelles)/(total micelle mass) × 100%; DLE (%) = (actual mass of loaded drug)/(initial drug input) × 100%.
Quantification of FITC-labeled CD44 targeting peptide on empty micelles, mP6 micelles, and mP6/Rg3 micellesEmpty micelles, mP6 micelles, and mP6/Rg3 micelles were incubated with 2 × 105 cells plated in 24-well culture plates at a final concentration of 0.5 μM for 30 min at 37 °C. Following two PBS washes, cells were fixed with 4% paraformaldehyde (8.18715, Sigma-Aldrich, USA) for 30 min at ambient temperature. Cells were centrifuged at 1000 rpm for 5 min using a ROTOFIX 32 A centrifuge (Hettich, Germany) and subjected to laser scanning confocal microscopy (OlympusFV1000, Center Valley, Pennsylvania). The CD44 targeting peptide was visualized with green fluorescence (FITC), while the cell nuclei were stained with blue fluorescence (DAPI).
Degradation of mP6/Rg3 micellesmP6/Rg3 micelles were suspended in PBS (pH 7.4) at 37 °C with continuous shaking. The initial dry mass (m₀) was recorded prior to degradation. At designated time points (0, 0.2, 0.3, 0.5, 1, 2, 3, 6, 8, 12, 14, 16, 18, 20, 22, and 24 days), samples were collected, freeze-dried, and weighed to determine mass loss (Δm). The degree of degradation was calculated based on the difference between the initial and residual dry mass.
Extracellular release of Rg3To investigate the release profile of Rg3 under physiological and tumor-mimicking conditions, mP6/Rg3 micelles (1 mL) were enclosed in dialysis membranes (MWCO 3.5 kDa) and immersed in 5 mL of PBS adjusted to pH 7.4, 6.5, or 5.0. Each setup was placed in a 50 mL centrifuge tube and incubated at 37 °C under mild agitation (100 rpm) in the absence of light. At scheduled intervals, 5 mL of the external buffer was collected and replenished with fresh PBS. The amount of Rg3 released into the buffer was determined by UV–visible spectrophotometry at 425 nm.
In vitro biocompatibility assessmentMurine Oral Mucosa Epithelial Cells (OMEC) (PCLM0161-RT, Bihe Biotech, China) were cultured in DMEM medium (12,430,047, Gibco, USA) containing 10% FBS (12,483,020, Gibco, USA) and 1% penicillin/streptomycin (15,070,063, Gibco, USA) at 37 °C with 5% CO₂.
Live/Dead Cell Staining: OMEC were plated in 12-well plates at 1 × 10⁶ cells/well and treated with 5 μM of empty micelles, mP6, or mP6/Rg3 for 24 h. Live/dead staining was performed using a mixture of PBS, calcein-AM, and propidium iodide (1 mL:3 μL:5 μL; ZY140632, Zeye Biotechnology, China). The mixture was introduced directly to the wells and cultured at 30 °C for 37 min. Fluorescence imaging was performed using an IMT-2 microscope (Olympus, Japan), with live and dead cells denoted by green and red fluorescence, respectively.
CCK8 Assay: WSU-HN30 cells (1 × 10⁶ cells/well) were exposed to different micellar formulations for 1, 2, or 3 days. CCK-8 reagent (100 μL/mL; C0037, Beyotime) was introduced and incubated for 2 h. Following incubation, 100 μL of supernatant was aliquoted into a 96-well microplate, and optical density at 450 nm was recorded utilizing the Synergy Neo2 Hybrid detection system (Agilent BioTek, USA).
Multicellular tumor spheroids (MCTS) modelWe established the MCTS model to enrich CSCs from the murine oral cancer cell line MOC2 (Bio-129558, biobw, China). Once adherent cultures reached 85–90% confluence, cells were dissociated into single-cell suspensions and resuspended in serum-free DMEM/F-12 medium (11,320,033, Gibco, USA) enriched with 2% B-27 (17,504,044, Gibco), 10 ng/mL EGF (AF-315–09-500UG, PeproTech), and 10 ng/mL bFGF (100-18C-1MG, PeproTech). cell suspension was diluted to a final density of 1 × 105 cells/mL and transferred onto poly(2-hydroxyethyl methacrylate)-coated dishes. Medium was refreshed every three days. On day 7, spheroids were harvested for subsequent analysis.
Spheroid formation experimentCells enriched by the MCTS model were plated onto ultra-low adhesion 96- or 6-well plates (Corning, MA, USA) and proliferated in DMEM/F12 medium enriched with 1% methylcellulose (9004–67-5, Sigma-Aldrich, USA), 1 × B-27, and 20 ng/ml each of EGF and bFGF. After two weeks of culture, cell morphology was examined utilizing an optical phase-contrast microscope (CX43, OLYMPUS, Japan), and cell diameter was measured utilizing LAS V4.9 software.
Transfection and groupingCSCs were maintained in DMEM/F12 medium (Gibco) enriched with 1% methylcellulose (Sigma-Aldrich), B27 (Invitrogen), and 20 ng/mL of both EGF and bFGF (Gibco). Lentiviruses carrying oe-ABCB1 (LM-2069, LMAI Bio, Shanghai) or control vector (oe-NC) were produced in HEK293T cells (Bio-72947, Beioubo Wei, Beijing) and packaged by Shengwu Bioengineering. For luciferase assays, HEK293T cells were co-transfected with Mock-luc or CFIm25-UP-luc constructs and helper plasmids utilizing Lipofectamine 2000(Thermo Fisher, USA). Lentiviral supernatants were collected, concentrated with a commercial reagent (631,231, Takara), and preserved at − 80 °C.
Lentiviral transduction was performed by seeding 5 × 105 CSCs into 6-well plates. At 70–90% confluency, cells were exposed to lentivirus (MOI = 10, ~ 5 × 10⁶ TU/mL) in the presence of 5 μg/mL polybrene (TR-1003, Sigma-Aldrich, UK) for 4 h. Medium was refreshed after dilution, and full replacement occurred at 24 h. Following 48 h of transduction, the luciferase signal was assessed, and transduced cells were selected with puromycin (E607054, Shengwu Bioengineering, Shanghai, China). Grouping schemes are detailed in Table S1.
Detection of EdU cells in cell by immunofluorescence assayThe 2',7'-dichlorofluorescein diacetate (DCFH-DA) assay kit (C0071S, Beyotime, China) was utilized to monitor EdU cells within cells. Nuclei were counterstained with Hoechst 33,342, and fluorescence signals were recorded utilizing an IMT-2 inverted fluorescence microscope (Olympus, Japan). The proliferation index was determined as the percentage of EdU-labeled nuclei relative to the total nuclei utilizing ImageJ software.
Scratch assay experimentConfluent CSC monolayers cultured in 6-well plates were subjected to a linear scratch using a sterile 200 μL pipette tip. Dislodged cells and debris were eliminated by rinsing three times with PBS. To minimize proliferation interference, cells were exposed to cytochalasin D (1 μg/mL; M5353, Sigma-Aldrich) for 1 h before scratching. Wound areas were imaged at 0, 24, and 48 h, and migration was quantified by calculating gap closure as a percentage of initial wound width. For each condition, five fields per well were imaged, with 6–10 regions randomly analyzed. All assays were conducted in triplicate.
Transwell experimentCell motility and invasive potential were assessed utilizing polycarbonate membrane inserts (8.0 μm pore size; CLS3422, Corning, USA) and Matrigel-coated invasion chambers (354,480, Corning). CSCs were seeded into the upper compartments containing DMEM enriched with 10% FBS. Non-migrated cells on the upper surface were removed with sterile swabs. The underside of the membrane was fixed in 4% paraformaldehyde and stained with crystal violet (C0121, Beyotime). Stained cells were visualized utilizing an inverted microscope (XDS-900, Caikon), with five regions per well imaged and 6–10 randomly selected fields analyzed per region. Quantitative analysis was conducted in ImageJ utilizing the"Analyze Particles"tool. Each experiment was performed in triplicate.
Flow cytometryDetection of CD44: CSCs were enzymatically dissociated with 0.25% trypsin solution (25200072, Gibco, USA) and subsequently diluted to 1 × 10⁶ cells in 200 μL of flow cytometry buffer (660585, BD Biosciences, USA). Cells were incubated with 2 μL of fluorophore-conjugated CD44 antibody (ab316123, 1:500, Abcam, UK) or isotype control on ice for 30 min. After staining, cells were washed, fixed in 10% formalin (R04587, Merck, USA), and analyzed on a BD FACSCalibur flow cytometer (BD Biosciences, USA).
Apoptosis detection: To assess apoptotic responses, Huh-7 cells (2 × 105 per well) were seeded in 6-well plates and collected with trypsin (R001100, Gibco, USA). Following centrifugation at 800 g, cells were reconstituted in 500 μL of binding solution provided in the Annexin V-FITC/PI apoptosis kit (556547, BD Biosciences, USA), followed by incubation with 5 μL Annexin V-FITC and 5 μL propidium iodide for 15 min at ambient temperature in the absence of light. Samples were then immediately subjected to flow cytometric assessment using a BD FACSCalibur system. Quadrant gating was applied to identify viable, early apoptotic, late apoptotic, and necrotic cells. The overall apoptotic rate was determined by combining the proportions of early and late apoptotic cell populations. All experiments were independently performed in triplicate.
Animal experiment in vivoSixty healthy male BALB/c nude mice (4–6 weeks old, Charles River, 401, China) were individually housed under SPF conditions. The animal facility was maintained at 22–25 °C with 60%–65% relative humidity and a 12 h light/dark cycle. Sterilized food and water were provided ad libitum. Animals were acclimated for one week prior to experimental procedures, and their health status was observed prior to the experiment.
To establish an OSCC model, 100 μl of MOC2 cells (2 × 106) were inoculated at the left edge of the tongues of the mice to develop the OSCC mouse model. This procedure was repeated twice a week for one week. After 4 weeks, bioluminescence imaging was performed utilizing IVIS Lumina Series III (PerkinElmer, USA). Mice were then euthanized with the intraperitoneal injection of pentobarbital sodium (100 mg/kg; 11715, Merck, USA). Tumors were excised, and their maximal diameter and weight were recorded for further analysis (Kioi et al. 2008).
To establish an OSCC lung metastasis model, 100 μl of MOC2 cells (2 × 106) were injected intravenously into the nude mice to induce the development of OSCC in the lungs. The injections were performed twice a week for one week. After four weeks, animals were euthanized as described above, and lung tissues were harvested for subsequent pathological evaluation.
All animal experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals (Jiang et al. 2020) and approved by the Institutional Animal Ethics Committee. Details of the mouse groups can be found in Table S2.
Hematoxylin and eosin (H&E) stainingMouse oral mucosa, tumor, and lung tissues preserved in formalin were embedded in paraffin, sectioned at 5 μm thickness, and sampled every 200 μm. Following xylene-mediated dewaxing and ethanol gradient rehydration, the slices were immersed in Harris hematoxylin (ST2067, Beyotime) for 5 min, differentiated in 0.5% acid alcohol (C0165S, Beyotime) for 10 s, and counterstained with eosin (C0109, Beyotime) for 40 s. After dehydration and clearing, slides were sealed with neutral resin. Tissue architecture and pathology were visualized utilizing bright-field microscopy.
TUNEL stainingApoptotic cells in mouse tumor tissues were detected utilizing a TUNEL staining kit (C1086, Beyotime, China). Paraffin-embedded sections were first treated with 3% hydrogen peroxide to block endogenous peroxidase activity, then incubated with 50 μL of TUNEL working solution at 37 °C for 1 h in darkness. Cells were counterstained with DAPI for 30 min to visualize nuclear DNA, then rinsed three times with PBS. Fluorescent signals were captured at 400 × magnification utilizing a BX53 epifluorescence microscope (Olympus, Japan). Apoptotic index was quantified in ImageJ by calculating the ratio of TUNEL-positive area to total nuclear staining. Each group contained five animals, with five paraffin sections prepared per animal and 6–10 randomly selected microscopic fields evaluated per section.
High-throughput transcriptome sequencingTotal RNA was isolated from normal oral mucosa and OSCC tumor tissues collected from the Normal (n = 3) and OSCC (n = 3) groups. Sequencing was conducted by CapitalBio Technology (Beijing, China). Total RNA (5 μg per sample) was subjected to ribosomal RNA depletion utilizing the Ribo-Zero Magnetic Kit (MRZG12324, Epicentre, USA). Strand-specific RNA-seq libraries were generated with the NEBNext Ultra RNA Library Prep Kit (E7760S, NEB, USA), including ~ 300 bp RNA fragmentation, random hexamer priming, and reverse transcription for cDNA synthesis. Subsequent steps involved end-repair, dA-tailing, and ligation of Illumina sequencing adapters. USER enzyme (M5508, NEB) was applied to preserve strand specificity. Libraries were PCR-amplified, assessed for quality using the Agilent 2100 Bioanalyzer, and quantified via the KAPA Library Quantification Kit (KK3605, Merck, USA). Paired-end sequencing was performed on the Illumina NextSeqCN500 platform.
Transcriptomic data processing and analysisRaw paired-end sequencing data were initially evaluated for quality utilizing FastQC (v0.11.8). Adapter contamination and poly(A) stretches were eliminated via Cutadapt (v1.18). Reads containing over 5% undefined bases (N) were filtered out through custom Perl scripts. The FASTX Toolkit (v0.0.13) was employed to retain only sequences with at least 70% of bases scoring above Q20. Paired-end integrity was restored utilizing BBMap. High-quality reads were aligned to the GRCh38 human genome reference utilizing HISAT2 (v0.7.12).
Differential expression was analyzed via the"edgeR"package in R, applying thresholds of |log₂ fold change|> 1 and adjusted p-value < 0.05. Shared DEGs were visualized as heatmaps utilizing the pheatmap package.
To identify genes with predictive significance, LASSO regression was conducted via the glmnet package. Optimal λ values were determined by cross-validation (cv.glmnet), and selection results were presented through coefficient trajectory plots.
A support vector machine (SVM) classifier was constructed utilizing the e1071 package with a radial basis function (RBF) kernel. Model tuning was carried out via grid search optimization of parameters C and γ (tune.svm). Feature refinement was implemented through Recursive Feature Elimination (RFE), iteratively discarding low-weight predictors.
Functional annotation of DEGs was conducted through Gene Ontology (GO) and KEGG pathway enrichment using clusterProfiler, org.Hs.eg.db, enrichplot, DOSE, and ggplot2 in R.
ROC curve construction and survival analysisTranscriptomic, clinical, and methylation data for 362 OSCC patients with bone marrow samples were obtained from TCGA (https://portal.gdc.cancer.gov/). ROC analysis was performed utilizing the R package “pROC” to evaluate the diagnostic performance of selected gene signatures in both training and validation cohorts. A risk score (RS) was calculated based on gene expression profiles and LASSO-derived coefficients. Based on the median RS, patients were grouped into high- and low-risk categories. Kaplan–Meier survival analysis was performed via the “survfit” function in R, and differences in overall survival were statistically tested utilizing the log-rank approach.
Quantitative proteomics analysisTumor tissues from OSCC mice (n = 6 per group) were lysed in RIPA buffer supplemented with protease inhibitors. Samples were sonicated three times (30 s every 5 min) to ensure efficient disruption and protein release. Protein concentrations were quantified via BCA assay (23,225, Thermo Fisher, USA), and standardized using the calibration curve. Following pH adjustment to 8.0, proteins were digested overnight at 37 °C with trypsin (enzyme:substrate ratio 1:50). Peptide desalting was performed using ZipTip C18 tips, and the samples were subjected to high-performance liquid chromatography (Shimadzu, Japan) coupled to a QSTAR Elite Hybrid mass spectrometer(MS) (Applied Biosystems/MDS-SCIEX). Each sample underwent triplicate technical runs. Peptide separation was performed on an in-house packed nano-C18 column (75 µm × 15 cm, 5 µm) using a 90-min linear gradient with mobile phases A (0.1% FA/2% ACN) and B (0.1% FA/100% ACN), flowing at 0.2 µL/min. MS detection was operated in positive ion mode within an m/z range of 300–2000. Ions with charges + 2 to + 4 and intensities > 5 were selected for fragmentation, applying a dynamic exclusion window of 30 s (tolerance: 30 mDa). Collision energy was automatically set during DDA acquisition, with a max ion accumulation time of 2 s. To expand protein identification, DIA (Data-Independent Acquisition) was also performed under the following settings: MS1 resolution at 120,000 (range: 350–1500 m/z, AGC: 4e5, injection time: 50 ms), MS2 resolution at 30,000 (AGC: 1e5, CE: 33 eV), and 47 overlapping windows calibrated with iRT standards. Raw data were analyzed using MaxQuant for DDA and Spectronaut V13 (Biognosys, Switzerland) for DIA, applying label-free quantification and data normalization. Differentially expressed proteins (DEPs) were determined via Welch’s ANOVA (P < 0.05, |log₂FC|> 1).
Metabolomics analysisOral normal mucosa and tumor specimens were harvested from mice in the Normal (n = 6), OSCC (n = 6), and mP6/Rg3-treated (n = 6) cohorts. For each sample, 300 μL of tissue homogenate was placed into 1.5 mL polypropylene tubes, extracted with 900 μL of 80% methanol containing 0.1% formic acid, vortexed for 2 min, and centrifuged at 12,000 g for 10 min. The resulting supernatant was transferred into autosampler vials for subsequent metabolomics profiling.
Chromatographic separation was performed on a Shimadzu LC-20 UPLC system coupled to an AB Sciex Triple TOF 6600 MS utilizing a Waters ACQUITY UPLC HSS T3 C18 column (100 × 2.1 mm, 1.8 μm, 40 °C; flow rate 0.4 mL/min). The mobile phases were 0.1% formic acid in water (A) and acetonitrile (B). The elution gradient was 5% B (0–11 min), 90% B (12 min), and back to 5% B (12.1–14 min). The column effluent was transferred into the MS without splitting.
MS parameters included an ion spray voltage of 5500 V, source temperature of 550 °C, and gas pressures of 50 psi (nebulizer) and 60 psi (auxiliary). OPLS-DA with 100 permutations evaluated model robustness. Differential metabolites (DMs) were identified based on VIP > 1 and p < 0.05, with further filtering by |fold change|≥ 2 or ≤ 0.5 (Student’s t-test). Metabolic pathway enrichment was conducted using MetaboAnalyst v5.0.
Expression of proteins detected by immunofluorescent stainingCSC cells were fixed in 4% paraformaldehyde at ambient temperature for 15 min and rinsed twice with PBS. Membrane permeabilization was performed using 0.5% Triton X-100 (P0096, Beyotime, China) for 10 min. For immunofluorescence, cells and tissue sections were incubated overnight at 4 °C with rabbit anti-ABCB1 (MA1-26528, 1:200), ABCA1 (PA1-16789, 1:500), ABCB11 (PA5-114800, 1:200), ABCC1 (MA5-16112, 1:500), and ABCD1 (11159–1-AP, 1:100; Proteintech). After thorough PBS washes, cytoskeletal structures were labeled with either FITC- or rhodamine-conjugated phalloidin (F432 or R415, Invitrogen) for 1 h, followed by Alexa Fluor 647- or 488-conjugated goat anti-rabbit IgG (ab150083 or ab150077, 1:200; Abcam) for another hour. Nuclei were counterstained with DAPI (D3571, 10 μg/mL) for 10 min at ambient temperature. Samples were stored at 4 °C until imaging. Fluorescent images were captured using an IMT-2 fluorescence microscope (Olympus, Japan). Fluorescence intensity was quantified in ImageJ as the ratio of target signal to nuclear staining. For each group, five sections per sample were analyzed, with 6–10 randomly selected fields evaluated per section. All experiments were repeated in triplicate.
Measurement of intracellular Fe2+ levelsTo determine intracellular ferrous iron (Fe2⁺) levels, CSC cells were first cultured on on confocal-compatible dishes and rinsed with Hank's Balanced Salt Solution (HBSS; 13150016, Gibco, USA). Cells were subsequently exposed to 1 μM FerroOrange (F374, Dojindo, Japan) for 30 min and visualized utilizing a laser scanning confocal microscope (LSM780, Zeiss, Germany). In addition, total iron levels in cellular and tissue lysates were quantified utilizing an iron assay kit (ab83366, Abcam).
TEMCSC samples were initially fixed with 2.5% glutaraldehyde at 4 °C overnight, followed by post-fixation with 1% osmium tetroxide for 1–2 h. Dehydration was performed at ambient temperature through graded ethanol (50–95%) and acetone series. Samples were embedded in resin and polymerized at 70 °C overnight. Ultrathin Sects. (70–90 nm) were cut utilizing a Reichert ultramicrotome and stained sequentially with uranyl acetate (50% ethanol-saturated) and lead citrate for 15 min each. Ultrastructural examination was conducted utilizing a TEM (TECNAI G2 F20, FEI, USA).
Mitochondrial membrane potential (MMP) detectionCSC cells were cultured in 6-well plates and stained with 1 mL of JC-1 working solution (40706ES60, YiSheng Biotechnology, China) at 37 °C for 20 min. For positive control, 50 μM CCCP was introduced at a 1:1000 dilution and treated under identical conditions. After staining, cells were rinsed twice with 1 × JC-1 buffer, then overlaid with 2 mL of phenol red- and serum-containing medium. JC-1 monomer fluorescence was detected utilizing either fluorescence or confocal microscopy (Ex: 490 nm, Em: 530 nm).
Biochemical assaysThe intracellular and tissue concentrations of reduced (GSH) and oxidized glutathione (GSSG) were assessed utilizing commercial quantification kits (S0053, Beyotime Biotechnology, China). Absorbance at 450 nm was recorded with a microplate spectrophotometer (Infinite200, Tecan, Beijing), and concentrations were calculated utilizing standard curves.
For intracellular reactive oxygen species (ROS) detection, cells seeded in 6-well plates were incubated with 10 μM DCFH-DA (S0033S, Beyotime, Jiangsu) for 20 min in the dark. After trypsinization and centrifugation, cells were resuspended and analyzed by flow cytometry (FC500ML, Beckman Coulter). To visualize ROS generation at the cellular level, an additional set was stained under the same conditions and imaged utilizing a confocal microscope (LSM780, Zeiss; Ex/Em: 488/525 nm). Given the suboptimal specificity of DCFH-DA toward H₂O₂, additional assessment of lipid oxidative damage was conducted to complement these measurements.
For ROS detection in mouse tumor tissues, the BH-02X9621 kit (Shanghai Bohu Biotechnology) was employed. Homogenized supernatants (200 μL) were mixed with 2 μL of dihydroethidium (DHE) probe in 96-well plates and incubated at 37 °C in a dark environment for 30 min. Following PBS rinses, nuclear counterstaining with DAPI, and slide mounting, fluorescence signals were quantified utilizing a microplate reader with excitation at 488–535 nm and emission at 610 nm.
To evaluate lipid peroxidation, cells were labeled with 5 μM C11-BODIPY581/591 (D3861, Invitrogen) for 15 min under light-protected conditions. Oxidation-driven red-to-green fluorescence shifts were observed microscopically. In addition, malondialdehyde (MDA) content was assessed utilizing a colorimetric kit (BC0025, Solarbio, Beijing). Absorbance at 600 nm, 532 nm, and 450 nm was recorded, and MDA concentrations were calculated accordingly.
Immunohistochemistry experimentTissue samples from mouse tumor and lung tissues were fixed in 4% paraformaldehyde, dehydrated, cleared, embedded in paraffin, and then sectioned. For immunohistochemical staining, tissue slices underwent deparaffinization and hydration, followed by heat-mediated epitope retrieval using microwave-assisted boiling in PBS. A polymer-based detection system (PV-9000, Proteintech, USA) was used for signal amplification. Sections were incubated overnight at 4 °C with primary antibodies diluted 1:500, including ABCB1 (ab170904), ABCA1 (ab18180), ABCB11 (ab315475), ABCC1 (ab260038), ABCD1 (ab124768), Ki67 (ab15580), N-cadherin (ab76011), E-cadherin (ab76319), and Vimentin (ab92547) (all from Abcam). Immunoreactive signals were visualized under a light microscope (CX43, Olympus, Japan), with positive staining identified by brown to yellow–brown chromogenic deposits. For quantification, five sections per mouse were analyzed in each group (n = 6 mice), and 6–10 fields per section were randomly selected. Semi-quantitative analysis of the stained regions was conducted utilizing ImageJ software.
Western blotTotal protein lysates were prepared from cultured cells and mouse tumor tissues using RIPA lysis buffer (P0013B, Beyotime, China), and concentrations were measured via BCA assay (A53226, Thermo Fisher, USA). Equal protein amounts were subjected to SDS-PAGE, followed by electrophoretic transfer onto PVDF membranes (PVH85R, Millipore, Germany) via a wet blotting system. After transfer, membranes were blocked with 10% BSA (37,520, Thermo, USA) at ambient temperature for 1 h, followed by overnight incubation at 4 °C with primary antibodies (1:1000, Abcam) targeting ABCB1 (ab170904), ABCA1 (ab18180), ABCB11 (ab315475), ABCC1 (ab260038), ABCD1 (ab124768), CD44 (ab243894), OCT4 (ab181557), NANOG (ab317506), SOX2 (ab92494), and the internal control GAPDH (ab9485). Following washes, membranes were incubated for 2 h with horseradish peroxidase-labeled goat anti-rabbit IgG (ab6721, 1:5000). Visualization of protein bands was achieved using a Syngene G:BOX F3 chemiluminescence imaging system (Antpedia, China), and signal intensity was quantified with ImageJ software. Expression levels were normalized to GAPDH and presented as grayscale ratios. Each cell experiment was conducted in triplicate, and animal experiments included six biological replicates per group.
RT-qPCRTotal RNA from cultured cells and tumor tissues was extracted using TRIzol reagent (ThermoFisher, USA), and its quality was evaluated with a Nanodrop 2000 spectrophotometer (ThermoFisher, USA). Complementary DNA (cDNA) was synthesized from the extracted RNA utilizing the PrimeScript RT Reagent Kit (Takara, Japan). Quantitative real-time PCR was subsequently conducted utilizing the Fast SYBR Green PCR Kit (ThermoFisher, USA) in triplicate, with GAPDH as the internal control. Relative gene expression was determined by the 2−ΔΔCt method. All assays were independently repeated three times. Primer sequences are listed in Table S3.
Statistical analysisStatistical analyses of the research data were conducted utilizing SPSS v21.0 (IBM, USA). Data are expressed as mean ± standard deviation (SD). Prior to hypothesis testing, assumptions of normality and homogeneity of variance were evaluated. Two-group comparisons employed independent t-tests, while multiple group comparisons were assessed via one-way or repeated measures ANOVA, as appropriate. A significance level of P < 0.05 was used to indicate statistical significance.
Comments (0)