
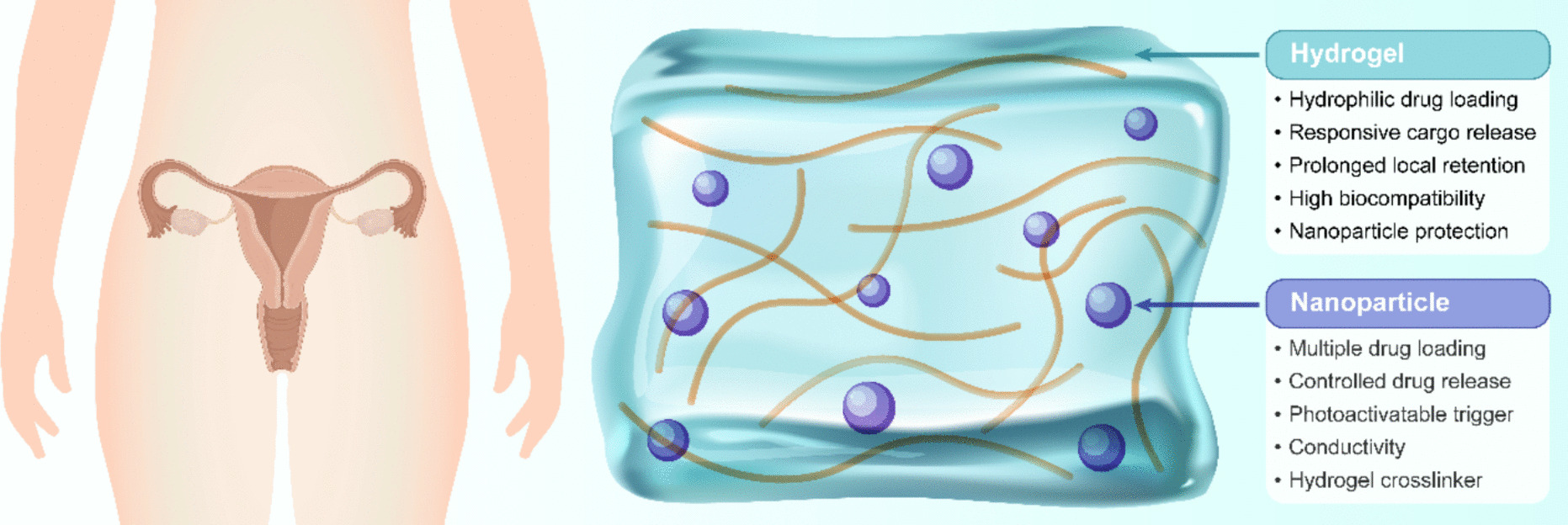
Remember me






GC is often late when you finally perceive its existence, and people are caught off guard (Norwood et al. 2022). What is more worrying being that the speed of GC progression is like a speeding train, and once diagnosed, the window for treatment is rapidly shortened. GC is regulated by a complex process involving multigene participation at multiple stages (Norwood et al. 2022). Etiological contributors such as Helicobacter pylori infection constituting the predominant risk factors associated with GC incidence (Norwood et al. 2022). Helicobacter pylori infection increases the m6A level in GC cells (Li et al. 2023a, b, c, d). Specifically, Helicobacter pylori infection extensively upregulates m6A regulators (METTL3, WTAP, FTO, ALKBH5) and downregulates key genes (PTPN14, ADAMTS1), linking m6A dysregulation to H. pylori-associated GC pathogenesis. Importantly, current studies indicate that the abnormal expression of METTL3, which operates as both an oncogene and a target gene, is involved in the onset and malignancies of GC.
METTL3 orchestrates multilayered oncogenic networks in GC pathogenesisMETTL3-mediated m6A modification, in conjunction with its interaction with m6A readers, is recognized as a crucial player in RNA processing, particularly in the regulation of mRNA dynamics, including their expression, stability, and eventual degradation, which is actively involved in GC development (Ding et al. 2023).
METTL3 as a bifunctional oncogenic hub Progression driverA comprehensive dataset analysis conducted by Liu et al. (2020) demonstrates a marked upregulation of METTL3 within GC tissues, in sharp contrast to the levels detected within healthy counterparts. Generally, METTL3 acts as the predominant m6A catalytic enzyme in the nucleus (Bedi et al. 2023). Notably, METTL3 also exhibits high cytoplasmic expression, which clinical studies have linked closely to GC progression (Wei et al. 2022). Their research uncovered an m6A-independent regulatory mechanism: cytoplasmic METTL3 associates with poly(A)-binding protein cytoplasmic 1 (PABPC1), strengthening the latter's interaction with the cap-binding complex eIF4F. This interaction selectively enhances the translation of oncogenic epigenetic factors (e.g., HDACs, DNMTs) without relying on m6A modification (Table 1). Importantly, such cytoplasmic METTL3-driven translational upregulation of epigenetic regulators contributes to carcinogenesis, highlighting its potential as a diagnostic and prognostic marker for GC patients.
Table 1 Key targets of METTL3 in GC pathogenesisThe expression level of METTL3 could be regulated in various manners. Transcription factors (TFs) are a class of sequence‐specific DNA‐binding proteins and are also an important class of proteins responsible for gene expression (Kawasaki and Fukaya 2023). Activating transcription factor 2 (ATF2), one member of the basic leucine zipper TF family, plays a role in chromatin remodeling, DNA damage response, transcriptional regulation, as well as tumor progression (Huebner et al. 2019). An elevated level of ATF2 is found in GC tissues and cell lines, which promotes GC cell growth and inhibits cell apoptosis, contributing to the short survival time in patients with GC. Meng et al. (2023a, b) found that ATF2 binds to METTL3’s promoter and enhances METTL3’s transcription. METTL3 in turn targets and increases cyclin D1 (CCD1) expression to promote cell cycle progression. These findings suggest that ATF2 is an upstream regulator for METTL3 expression and facilitates GC progression via activating the METTL3/CCD1 signaling pathway. Therefore, the oncogenic role of METTL3 is promoted by ATF2, suggesting intervention of METTL3 might be an anti‐tumor method for GC. In line with this, HOXA10 plays an essential role in tumor progression (Song and Zhou 2021). The importance of HOXA10 becomes evident in its strategic localization within the promoter region of transforming growth factor β2 (TGF-β2), stimulating its transcription and enhancing its secretion. HOXA10 overexpression upregulates METTL3 and global m⁶A levels through activating the TGF-β2/Smad cascade, with Smad proteins critically mediating METTL3 expression. Both HOXA10 and METTL3 hold clinical significance; notably, METTL3 underlies HOXA10-driven EMT progression (Song and Zhou 2021). These findings collectively underscore the essential role of the HOXA10-TGFβ2/Smad-METTL3 axis in promoting GC progression (Fig. 1, Table 1).
Fig. 1
METTL3 induces GC development and predicts poor outcomes. Targets, mechanisms, and functions of METTL3 in GC are comprehensively summarized and analyzed. METTL3 functions as an oncogene or regulates target genes involved in the process of apoptosis, aerobic glycolysis, angiogenesis, and immune microenvironment of GC, contributing to tumorigenesis, EMT progression, metastasis, and malignant phenotypes. Methyltransferase-3 (METTL3). N6-methyladenosine (m6A). Gastric cancer (GC). Basic leucine zipper ATF-like transcription factor 2 (BATF2). Elevated sphingosine kinase 2 (SPHK2). N.6-Methyladenosine RNA binding protein (YTHDF). Insulin-like growth factor 2 mRNA binding protein (IGF2BP). Suppressor of variegation 3–9 homolog 2 (SUV39H2). Epithelial-mesenchymal transition (EMT). Heparin-binding growth factor (HDGF). Hepatitis B X-interacting protein (HBXIP). C-Myc proto-oncogene (MYC). Homeobox A10 (HOXA10). Transforming growth factor β (TGF-β). Protein phosphatase 2 catalytic subunit alpha (PP2Acα). Cyclin D1 (CCD1). Phosphatase and tensin homolog (PTEN). Angiopoietin-like 3 (ANGPTL3). CUB domain-containing protein 1 (CDCP1). Epstein–Barr virus (EBV)
The escalation of METTL3 expression exhibits a positive correlation with the progression of tumor stage and grade, along with poor survival outcomes (Liu et al. 2020). Consequently, METTL3 emerges as an unfavorable prognostic factor in patients with GC. Further, METTL3 deletion reduces m6A modification on Gfi1 mRNA, decreasing its stability and downstream α-SMA expression, which suppresses EMT-driven proliferation and migration (Liu et al. 2020). Crucially, Gfi1 knockout in METTL3-WT cells phenocopied METTL3 loss, while Gfi1 overexpression rescued METTL3-deficiency effects (Liu et al. 2020). This intricate molecular cascade imparts constraints upon cellular proliferation and migration capacity. These findings collectively reveal that METTL3 acts as an oncogenic driver in GC by regulating Gfi1 expression (Fig. 1,Table 1).
Subsequent studies have further clarified METTL3's promotive role in regulating GC cell proliferation and migration, which operates via m6A modification of tumor-related genes—including YAP1 (Zhou et al. 2021), SOCS2 (Jiang et al. 2020), and basic leucine zipper ATF-like transcription factor 2 (BATF2) mRNA (Xie et al. 2020). BATF2 is upregulated in GC tissues and correlates with tumor progression. Consistently, Xie et al. (2020) revealed that BATF2 stabilizes p53 while inhibiting ERK phosphorylation, thereby driving GC progression through the METTL3-BATF2/p53-ERK axis. BATF2 knockdown abolished METTL3-driven proliferation, confirming BATF2 as a functional effector (Xie et al. 2020). Consistently, a substantial upregulation of hepatitis B X-interacting protein (HBXIP) relates to GC malignancies. Yang et al. (2020a, b) demonstrated that METTL3, as a target of HBXIP, upregulates MYC and its target glycolytic genes (MCM5, MCM6), linking m6A modification to metabolic reprogramming. Similarly, an elevated level of sphingosine kinase 2 (SPHK2) facilitates the malignancy properties of GC cells, indicating an unfavorable outcome (Huo et al. 2021). In elucidating this intricate interplay, they demonstrated that METTL3 coordinates with YTHDF1 to facilitate SPHK2 translation, which subsequently promotes KLF transcription factor 2 (KLF2) phosphorylation. The resulting ubiquitination and degradation of the KLF2 protein ultimately inhibits KLF2 expression in GC, thereby amplifying the manifestation of these aggressive traits. Similarly, Huo et al. (2021) reported that METTL3/YTHDF1 enhance SPHK2 translation, which phosphorylates and degrades KLF2, promoting proliferation and migration. In METTL3-knockout GC cells, re-expression of SPHK2 (via lentiviral transduction) restored proliferation and migration, confirming SPHK2 as the functional effector of METTL3 (Huo et al. 2021). Therefore, METTL3 functions by targeting the SPHK2/KLF2 axis. (Fig. 1, Table 1).
During tumor progression, the DNA damage repair mechanism can be reprogrammed by influencing the genomic integrity (Kiwerska and Szyfter 2019). A high level of suppressor of variegation 3–9 homolog 2 (SUV39H2) is detected in GC tissues and cell lines, which promotes proliferation and inhibits apoptosis in GC cells (Yang et al. 2023). Moreover, SUV39H2 acts as a histone methyltransferase that induces ataxia-telangiectasia mutated phosphorylation (ATM) phosphorylation by inhibiting DUSP6 transcription, which finally promotes homologous recombination and inhibits GC chemo-sensitivity to Cisplatin. Yang et al. (2023) found that METTL3/IGF2BP2-dependent m6A modification on SUV39H2 mRNA enhances SUV39H2 mRNA stability and upregulates SUV39H2 expression. Crucially, SUV39H2 knockout in GC cell lines reversed METTL3-driven chemoresistance, confirming SUV39H2 as the functional mediator of METTL3 in cisplatin resistance (Yang et al. 2023). Notably, METTL3 promotes homologous recombination and impairs the chemo-sensitivity of GC to Cisplatin by regulating SUV39H2 (Fig. 1, Table 1). Therefore, METTL3 with the expectation to be used as a target for GC precision therapy.
Metastatic switchThe elevated METTL3 expression shows a close correlation with an unfavorable prognosis in patients with GC (Okugawa et al. 2023; Ge et al. 2023; Yue et al. 2019). Yue et al. (2019) found an underlying mechanism wherein METTL3 enhances the stability of ZMYM1 in a HuR-dependent m6A modification pathway, which recruits the CtBP/LSD1/CoREST complex to repress the E-cadherin promoter and facilitate EMT and metastasis. Additionally, Wang et al. (2020) reported an intricate regulatory mechanism, whereby P300 activates METTL3 transcription by acetylating its promoter region with H3K27. Further, the activated METTL3, collaborates with IGF2BP3 recognition and combination on the modified site of the HDGF mRNA directly, resulting in an m6A modification on HDGF mRNA to promote its protein stability. Owing to the METTL3/IGF2BP3-mediated m6A modification on HDGF mRNA stability, HDGF protein production is increased in the cytoplasm, thereby promoting tumor angiogenesis. Concurrently, nuclear HDGF activates the expression of GLUT4 and ENO2, consequently promoting glycolysis in GC cells (Wang et al. 2020). In vivo, METTL3-knockdown xenografts showed reduced lung metastasis, which was reversed by HDGF overexpression (Wang et al. 2020). This process is closely associated with tumor growth and liver metastasis. Consequently, these findings suggest that elevated levels of METTL3 function as oncogenes that facilitate the development of GC by targeting IGF2BP3/HDGF or promote glycolysis by targeting the IGF2BP3/HDGF-GLUT4 and IGF2BP3/HDGF-ENO2 axes. In line with this, PLAGL2 is a zinc finger protein transcription factor that activates downstream targets, which is abnormally expressed in GC and enhances the proliferation and metastasis of GC. Chen et al. (2023) demonstrated that PLAGL2 binds to the upstream promoter of UCA1, which subsequently sponges miR-145-5p that targets YTHDF1. Moreover, YTHDF1 recognizes METTL3-mediated m6A on Snail by interacting with eEF-2 and thus promotes Snail expression, which eventually induces EMT and metastasis. Overall, PLAGL2 enhances GC progression via targeting Snail indirectly regulated by METTL3, suggesting that PLAGL2 is an upstream regulator for METTL3 expression and METTL3 is a therapeutic target for GC (Fig. 1,Table 1).
PBX1 functions as a positive transcriptional regulator of GCH1, enhancing its expression. Liu et al. (2022a, b) demonstrated that METTL3 interacts with PBX1 to stabilize its mRNA, which in turn induces GCH1 transcription and expression. The METTL3-PBX1-GCH1 axis elevates tetrahydrobiopterin (BH4) levels in GC cells, thereby promoting GC growth and lung/lymph node metastasis. Additionally, METTL3 deletion impairs xenograft tumor growth and lung/lymph node metastasis in vivo. Furthermore, DEK mRNA exhibits higher m6A modification levels in GC tissues and cell lines, which correlates with GC lung metastases (Zhang et al. 2022a, b). Mechanistically, METTL3 targets the 3'-UTR of DEK mRNA to install m6A modifications, thereby boosting DEK stability. In DEK knockout mouse models, lung metastasis induced by METTL3 overexpression was completely abolished, demonstrating DEK's essential role in METTL3-mediated metastasis (Zhang et al. 2022a, b). This study hints that METTL3 is the regulator of DEK function in GC lung metastasis. Therefore, METTL3 is a therapeutic target for inhibiting GC lung metastasis (Fig. 1,Table 1).
Furthermore, TGF-β/Smad signaling shows dual capacity as a tumor inhibitor and driver (Hu et al. 2018). METTL3 synergizes with the TGF-β/Smad pathway to promote the growth and metastasis of GC cells. In GC, evidence also demonstrates that the TGF-β/Smad2/3 axis plays the driver role in tumorigenesis and metastasis (Yuan et al. 2023). Mechanically, Yuan et al. (2023) reported that METTL3/IGF2BP2-dependent m6A modification stabilizes Smad3 mRNA and enhances Smad3 protein expression, which in turn activates the TGF-β/Smad pathway. Importantly, METTL3 interacts with p-Smad3 to regulate the transcription of downstream genes. These data suggest a novel regulation mechanism for the cancer-promoting function of the TGF-β/Smad3 signaling, for which METTL3 serves as the manipulator and might be a new therapeutic target for GC treatment (Fig. 1,Table 1).
Collectively, METTL3 is the key contributor to GC growth and metastasis. Therefore, targeting METTL3 might be therapeutic for GC.
Metabolic reprogrammingA strong correlation has been established between the diminished expression of protein phosphatase 2 catalytic subunit alpha (PP2Acα) and the manifestation of GC. Cheng et al. (2021) demonstrated that inhibiting PP2Acα activates ATM activity, leading to the upregulation of METTL3 and promoting the aggressive behavior of GC. Jointly, METTL3 acts as the target of the oncogene, PP2Acα, which functions by regulating the ATM/METTL3 axis. Similarly, another study indicated that METTL3 overexpression considerably amplifies its oncogenic function by mediating m6A on the MYC target genes, including MCM5 and MCM6 (Yang et al. 2020a, b). Moreover, NDUFA4 is overexpressed in GC, with its high levels linked to poor patient prognosis. Xu et al. (2022a, b) clarified that the METTL3-IGF2BP1 complex enhances m6A modification of NDUFA4 mRNA, thereby upregulating NDUFA4 in GC cells. This upregulation accelerates glycolytic and oxidative metabolism, triggering abnormal cell proliferation and tumor growth. Notably, inhibiting mitochondrial fission can reverse NDUFA4-mediated metabolic changes and tumor progression in GC. NDUFA4 knockout in GC cells reversed METTL3-induced glycolytic flux and tumor growth in xenograft models, providing direct evidence of NDUFA4 as a metabolic executor of METTL3 (Xu et al. 2022a, b). This study suggests that the METTL3-NDUFA4 axis leads to GC development (Fig. 1,Table 1).
Collectively, METTL3, m6A modified-SPHK2, and m6A modified-NDUFA4 are risk factors for inducing GC malignant phenotype, suggesting their potential for GC treatment.
Tumor suppressor silencing via m6A-epigenetic crosstalk ADAMTS9-PI3K/AKT suppression: YTHDF2-mediated mRNA decayA low level of ADAMTS9 is considered an independent prognostic factor for GC. Wang et al. (2022a, b, c) suggested that METTL3/YTHDF2-mediated m6A modification on ADAMTS9 promotes ADAMTS9 degradation and reduces ADAMTS9 level in GC, which subsequently triggers PI3K/AKT pathway and enhances tumor cell proliferation and angiogenesis, thereby promoting GC progression. Notably, METTL3 promotes GC progression by inhibiting the anti-oncogene ADAMTS9 and regulating the ADAMTS9-PI3K/AKT pathway (Fig. 1).
Angiopoietin-like 3 (ANGPTL3) inactivation: m6A-dependent translational blockadeThe Cancer Genome Atlas (TCGA) data found that METTL3 is frequently elevated in GC (Zhang et al. 2023). High expression level of METTL3 is more likely indicate advanced tumor node metastasis. On the other hand, METTL3 deletion effectively impedes the higher oncogenic capacity of GC, as reflected by slowed cell growth and diminished migration and invasion capacities. Importantly, the effect of METTL3 deletion is similar to ANGPTL3 enrichment that hinders the growth and metastasis of GC cells, whereas this effect is reversed partially by ANGPTL3 inhibition. Further exploring of the TCGA dataset found the co-expression of ANGPTL3 and METTL3 in GC. Specifically, METTL3-mediated m6A modification on ANGPTL3 mRNA decreases ANGPTL3 expression, which shortens the life span of patients with GC (Zhang et al. 2023). ANGPTL3 overexpression in METTL3-high GC cells significantly suppressed tumor growth in mouse xenografts (tumor volume reduced by 62%, p < 0.01), functionally validating ANGPTL3 as a tumor suppressor downstream of METTL3 (Zhang et al. 2023). Overall, these findings discover the METTL3-ANGPTL3 axis and its effect on GC malignant development, suggesting that METTL3 exerts the oncogenic role in GC by suppressing the anti-oncogene ANGPTL3 in an m6A-dependent manner, which contributes to the mechanism understanding of GC development (Fig. 1,Table 1).
SRSF11 splicing dysregulation: connects p53/apoptosis pathway impairmentA low level of SRSF11 is associated with poor survival in patients with GC, which is regulated by METTL3 (Oh et al. 2023). Specifically, METTL3 regulates SRSF11 mRNA splicing and lowers SRSF11 expression in patients with poor prognosis (Oh et al. 2023). According to gene set enrichment analysis, the downregulated level of SRSF11 relates to the pathways of p53/apoptosis, inflammation/immune response, and ultraviolet/reactive oxygen species stimulus–response in GC (Oh et al. 2023) (Fig. 1). These findings suggest that SRSF11-mediated poor prognosis in patients with GC relates to METTL3 functions. However, the exact signaling pathways are unclear and need to be clarified.
ncRNAs circuits amplify METTL3 oncogenicitymiRNA-m6A feedback loopsElevated levels of m6A and METTL3 are detected in GC, demonstrating associations with unfavorable prognoses and increased malignancy (Sun et al. 2020a, b). Beyond its established role in regulating mRNA, METTL3 functions in GC development by regulating ncRNAs, such as miRNAs. Dysregulation of miRNAs is associated with the progression of numerous tumors.
OncomiR activation: DGCR8-dependent pri-miR-17–92 processingA previous study reported that METTL3-mediated m6A modification on pri-miR-17–92 promotes miR-17–92 cluster formation through an m6A/DGCR8- dependent mechanism in GC (Sun et al. 2020a,
Comments (0)