Technological and computational advances in recent years, from cryo-electron microscopy to sequencing technologies and machine learning, have substantially deepened our understanding of RNA splicing. Nature Reviews Genetics and Nature Reviews Molecular Cell Biology present an online collection that showcases the biological insights facilitated by these advances.
The discovery of ‘split genes’ in 1977, for which Richard Roberts and Phillip Sharp were awarded the 1993 Nobel Prize in Physiology or Medicine, is just one example of many that illustrate the complexity of the flow of genetic information. In contrast to bacterial genes, where the linear sequence of nucleotides in a stretch of DNA corresponds directly to the linear sequence of amino acids in the protein, most eukaryotic genes are organized as a “mosaic”1: introns, sequences that are removed from the transcribed mRNA precursor, are interleaved with exons, sequences that are stitched together to produce the mature mRNA in a process known as RNA splicing. The spliceosome, a dynamic ribonucleoprotein complex composed of five small nuclear RNAs (snRNAs) and numerous associated proteins, recognizes conserved sequence motifs at exon–intron boundaries and carries out a two-step transesterification reaction to remove introns and ligate exons. Recent cryo-electron microscopy (cryo-EM) studies have yielded high-resolution structures of several conformational states of the spliceosome, revealing the dynamic rearrangements that drive intron removal and exon ligation2.
Splicing is essential for the accurate translation of DNA sequence information and comes with the added perk of generating transcriptomic and proteomic diversity in the form of alternative splicing — that is, the regulated inclusion or exclusion of exons. Alternative splicing greatly expands the coding potential of the genome; more than 95% of human multi-intron genes undergo alternative splicing, producing mRNA isoforms that can differ in coding sequence, regulatory elements or untranslated regions. These isoforms can influence mRNA stability, localization and translation output, thereby modulating cellular function.
“Splicing is essential for the accurate translation of DNA sequence information”
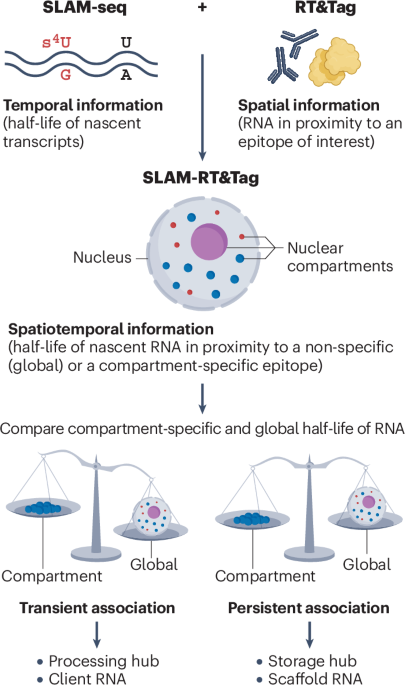
The timing of splicing relative to transcription has a substantial impact on alternative splicing. Splicing can occur either co-transcriptionally3, as the pre-mRNA is being synthesized, or post-transcriptionally4, after transcription is completed. Recent advances in long-read sequencing and imaging methods have provided insights into the timing and regulation of splicing, revealing its dynamic interplay with transcription and RNA processing. Moreover, it is now clear that splicing depends on the compartmentalization of its machinery and substrates within the nucleus — particularly within biomolecular condensates and membraneless organelles such as nuclear speckles and paraspeckles5. These structures, which arise by phase separation, provide dynamic environments that concentrate, organize and regulate splicing factors.
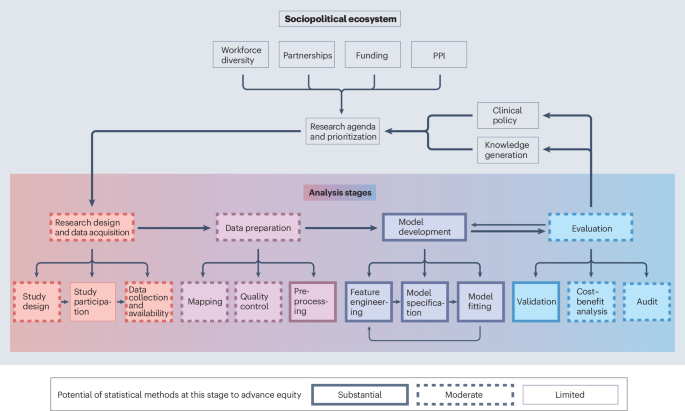
Mutations in core spliceosomal proteins can compromise splicing fidelity or efficiency. Genetic variation can also disrupt splice sites or regulatory elements, leading to exon skipping, intron retention or the activation of cryptic splice sites. Targeting RNA splicing with therapeutics, such as antisense oligonucleotides or small molecules, has emerged as a powerful and increasingly validated strategy to treat a range of diseases, particularly genetic disorders, neurodegenerative diseases and certain cancers1,2. Moreover, machine learning models are improving our ability to predict the effects of genetic variants on splicing6, with the potential to guide drug development and clinical diagnostics.
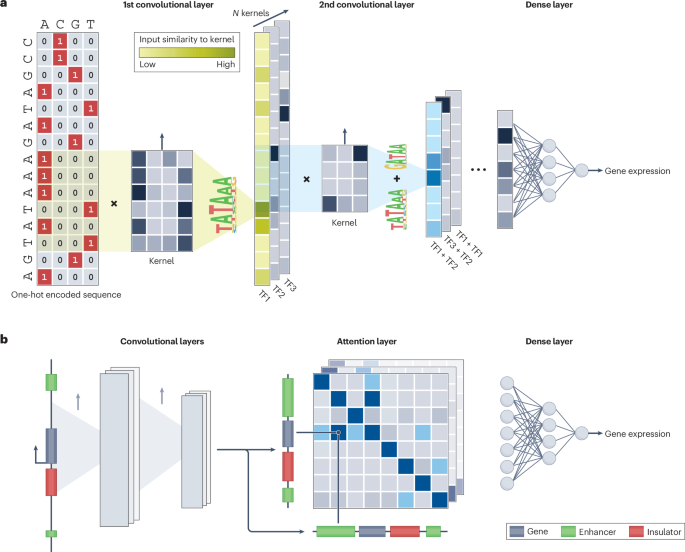
Deciphering the ‘splicing code’ has been a major challenge in computational biology and molecular genetics. The splicing code refers to “a set of rules, an algorithm or a computational model that could predict the splice isoforms, and their frequencies, produced from any transcribed gene in a specific cellular context”6. It encompasses cis-regulatory elements, RNA structure and trans-acting contributors to splicing regulation, which together determine which mRNA isoforms are produced. With the advent of large-scale transcriptomic and functional genomics data, machine learning approaches, particularly deep learning, have become powerful tools for modelling the splicing code, although interpretability remains a challenge.
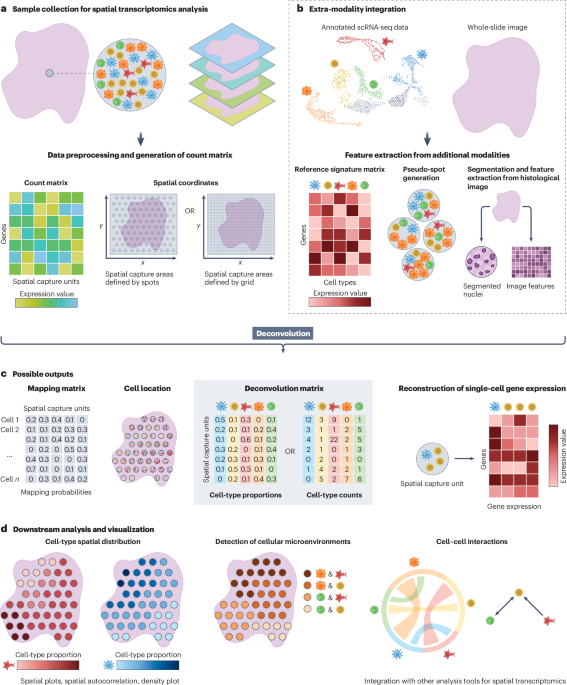
The ability of a single gene to produce several, functionally distinct protein isoforms through alternative splicing could enable organisms to rapidly adapt to changing environments. By enabling the sequencing of full-length transcripts, long-read sequencing data have yielded a more complete picture of alternative splicing. Subsequent comparative transcriptomic studies have revealed striking differences in the extent of alternative splicing between eukaryotes. Indeed, recent studies suggest that heritable variation in patterns of alternative splicing contributes to adaptive evolutionary change7.
Together, a large body of work has shed light on how splicing contributes to development, disease and evolutionary innovation. With increasing insights into the diversity, regulation and structural basis of splicing, researchers are poised to further decode how splicing shapes phenotypic outcomes and contributes to human disease.

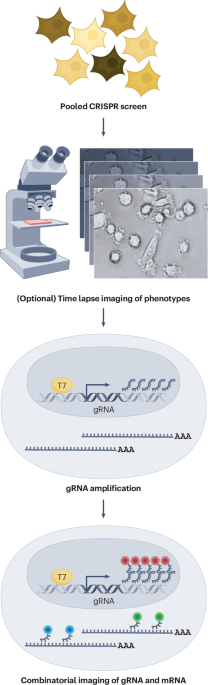
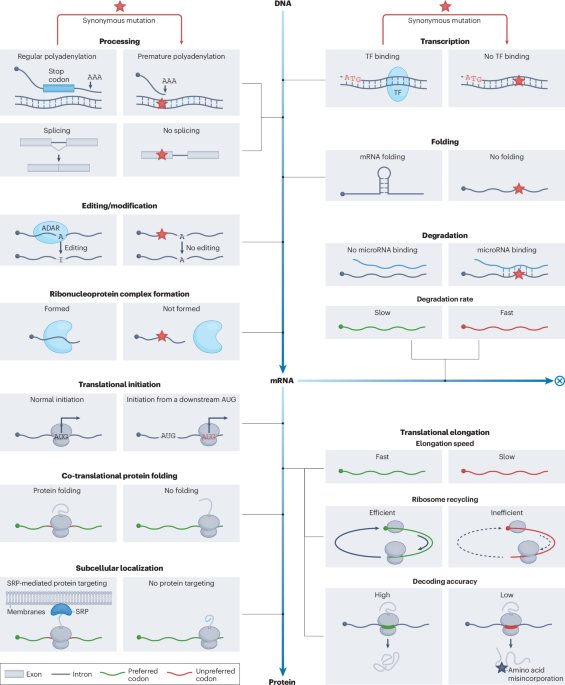

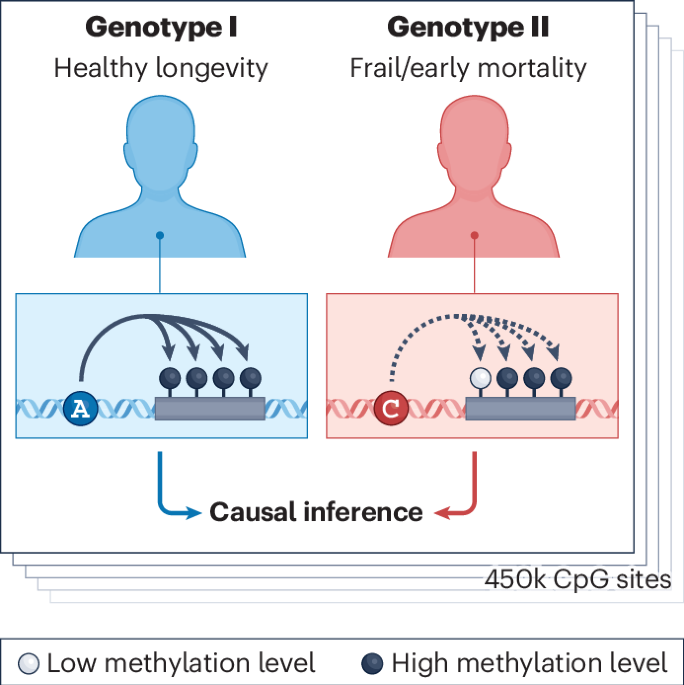
Comments (0)