
Remember me
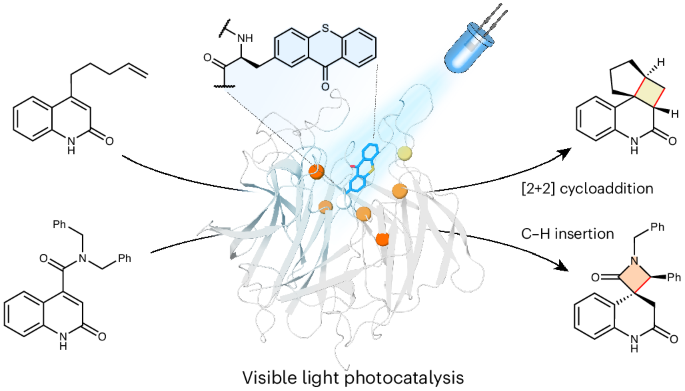


Substrates and products were chemically synthesized according to procedures reported in the ‘Chemical procedures’ section of the Supplementary Information. All other chemicals and biological materials were obtained from commercial suppliers. Lysozyme, DNase I, chloramphenicol and kanamycin were purchased from Sigma-Aldrich. Polymyxin B sulfate was obtained from Alfa Aesar. LB agar, LB media, 2× YT media, isopropyl-β-d-1-thiogalactopyranoside (IPTG) and arabinose were purchased from Formedium. The Mj aminoacyl tRNA synthetase in the plasmid pBK, named pBK_A9/tRNACUA41, was provided by E. J. Peterssen (University of Pennsylvania) and R. A. Mehl (Oregon State University). Escherichia coli strains BL21(DE3) and 5α, Q5 DNA polymerase, T4 DNA ligase and restriction enzymes were purchased from New England Biolabs. Oligonucleotides and genes were synthesized by Integrated DNA Technologies. Irradiation times given below refer to the total length of light exposure.
Synthetase engineeringConstruction of pEVOL_AcdRS2b A9 and engineered variantsThe pBK_ AcdRS2b_A9/tRNACUA plasmid41 was provided by E. J. Peterssen (University of Pennsylvania) and R. A. Mehl (Oregon State University). Two copies of the gene were cloned into the Mj pEVOL vector using BglII/SalI and NdeI/PstI restriction sites to yield the plasmid pEVOL_AcdRS2b_A9. The vector also contained the Mj tRNACUA. Evolved mutants of pEVOL_AcdRS2b_A9 were constructed using the same procedure after each round of synthetase engineering.
Synthetase library preparationLibraries were based on the engineered MjTyrRS pBK_ AcdRS2b_A9/tRNACUA41 (in pBK plasmids, under the control of E. coli GlnRS promoter and terminator on pBR322-derived kanamycin-resistant plasmids). Positions were individually randomized using degenerate NNK codons. DNA libraries were constructed by overlap extension polymerase chain reaction (PCR). Primers for library generation are given in Supplementary Table 12. Assembled genes and pBK vector were digested using NdeI and PstI endonucleases, gel-purified and subsequently ligated using T4 DNA ligase in a 5:1 ratio, respectively. Ligations were transformed into E. coli DH10b, the resulting colonies were pooled together and plasmid DNA was extracted using a Miniprep Kit (QIAGEN) to yield plasmid DNA for each library. Sequencing was performed by Source BioScience.
For protein expression and screening, all transfer and aliquoting steps were performed using Hamilton liquid-handling robots. Chemically competent E. coli DH10b cells harbouring pALS-GFP-TAG150 (containing an sfGFP reporter with a TAG codon at residue 150 and tyrosyl-tRNACUA) were transformed with the appropriate library plasmids. Freshly transformed colonies were used to inoculate 150 μl of defined non-inducing medium (Supplementary Information) supplemented with 50 μg ml−1 kanamycin and 25 μg ml−1 tetracycline in Corning Costar 96-well microtitre round-bottom plates. Each plate contained six freshly transformed clones of the parent template as internal references. Plates were incubated overnight at 30 °C, 80% humidity in a shaking incubator at 900 rpm. For each library plate, 20 μl of overnight culture was used to inoculate two separate deep-well blocks containing 480 μl of defined autoinducing medium (‘Materials’) supplemented with 50 μg ml−1 kanamycin, 25 μg ml−1 tetracycline, with one of the deep-well blocks containing no amino acid and the second deep-well block containing 0.5 mM m-TX amino acid (see chemical procedures for synthesis of m-TX amino acid in Supplementary Information) which was added as a 0.5-M stock dissolved in 1 M NaOH. The cultures were incubated at 30 °C, 80% humidity and 900 rpm for 48 h. Cells were gathered by centrifugation at 2,900g for 5 min. The supernatant was discarded and the pelleted cells were resuspended in 400 μl of PBS lysis buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4, 1.0 mg ml−1 lysozyme, 0.5 mg ml−1 polymyxin B and 1 μg ml−1 DNase I) and incubated for 2 h at 30 °C, 80% humidity, with shaking at 900 rpm. Cell debris was removed by centrifugation at 2,900g for 5 min. The clarified cell lysate was transferred to a 96-well microtitre plate (Costar) and fluorescence measurements were performed with excitation at 395 nm and emission at 509 nm using a BMG LabTech CLARIOstar spectrophotometer. The most active variants were rescreened as purified proteins and non-canonical amino acid incorporation verified by mass spectrometry (see below). Proteins were expressed and purified as described below with the exception that starter cultures were inoculated from glycerol stocks prepared from the original library plate overnight cultures. The engineered MjTyrRS pBK_mTX/tRNACUA (which contained the mutation L108W when compared to pBK_ AcdRS2b_A9/tRNACUA) was subcloned into pEVOL as described above and is now named pEVOL_mTX-RS.
Protein production and purificationChemically competent E. coli DH10b cells harbouring pALS-GFP-TAG150 (containing an sfGFP reporter with a TAG codon at residue 150 and tyrosyl-tRNACUA) were transformed with the appropriate pBK plasmid. A single colony of freshly transformed cells was cultured for 18 h in 10 ml defined non-inducing medium (Materials) containing 50 μg ml−1 kanamycin and 25 μg ml−1 tetracycline. Starter culture (0.1 ml) was used to inoculate 10 ml defined autoinducing medium (Materials) supplemented with 50 μg ml−1 kanamycin and 25 μg ml−1 tetracycline and 10 ml defined autoinducing medium (Materials) supplemented with 50 μg ml−1 kanamycin, 25 μg ml−1 tetracycline and 0.5 mM m-TX which was added as a 0.5-M stock dissolved in 1 M NaOH. Cultures were grown at 30 °C, 180 rpm for 48 h. Cells were subsequently collected by centrifugation (3,220g for 10 min). Pelleted cells were resuspended in lysis buffer (50 mM HEPES, 300 mM NaCl, pH 7.5 containing 20 mM imidazole) and lysed by sonication (1 s on/off, 5 min total sonication) at 4 °C with the addition of 1 µg ml−1 DNase I. Cell lysates were clarified by centrifugation (27,216g for 30 min) and supernatants were subjected to affinity chromatography using Ni-NTA Agarose (QIAGEN). 6-His-tagged proteins were eluted using 50 mM HEPES, 300 mM NaCl, pH 7.5 containing 250 mM imidazole. Buffer exchange of purified proteins was performed using 10DG desalting columns (Bio-Rad) and phosphate-buffered saline (PBS) pH 7.4 (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4) and analysed by mass spectrometry (see below).
Photoenzyme engineeringConstruction of pET-29b_VEnT1.0, pET-29b_SpEnT1.0 and variantsThe original DA_20_00 design43 was subcloned using NdeI and XhoI restriction sites into a pET-29b(+) vector modified to include a Strep-tag before the XhoI restriction site to yield pET-29b(+)_DA_20_00_Strep. The Ala173mTX mutation was introduced by replacing the Ala173 codon with a TAG stop codon using QuikChange site-directed mutagenesis (Agilent) to yield pET-29b(+)_VEnT1.0. To construct pET-29b(+)_SpEnT1.0, a Trp244mTX mutation was introduced by replacing the Trp244 codon with a TAG stop codon using QuikChange site-directed mutagenesis (Agilent). Point mutants of VEnT1.0 and SpEnT1.0 were constructed using the same procedure.
Protein production and purificationFor expression of VEnT1.0, SpEnT1.0 and their variants, chemically competent E. coli BL21(DE3) cells containing pEVOL_mTX-RS/tRNACUA were transformed with the appropriate pET-29b(+) construct. A single colony of freshly transformed cells was used to inoculate 5 ml LB medium containing 50 μg ml−1 kanamycin and 25 μg ml−1 chloramphenicol and cultured for 18 h at 37 °C and 200 rpm. Starter cultures (500 µl) were used to inoculate 50 ml 2× YT medium supplemented with 50 μg ml−1 kanamycin, 25 μg ml−1 chloramphenicol and 0.5 mM 2-amino-3-(9-oxo-9H-thioxanthen-2-yl)propanoic acid (mTX; Chemical procedures in Supplementary Information) or 0.5 mM 2-amino-3-(3-benzoylphenyl)propanoic acid (mBpA; Chemical procedures in Supplementary Information), which was added as a 0.5-M stock dissolved in 1 M NaOH. Cultures were grown at 37 °C, 200 rpm to an optical density at 600 nm (OD600) of about 0.6 a.u. Protein expression was induced with the addition of l-arabinose to a final concentration of 0.05% and IPTG to a final concentration of 0.1 mM. Induced cultures were incubated for 20 h at 25 °C and the cells were subsequently collected by centrifugation (3,220g for 10 min). (1 s on/off, 5-min total sonication) at 4 °C with the addition of 1 µg ml−1 DNase I. Cell lysates were clarified by centrifugation (27,216g for 30 min) and supernatants were subjected to affinity chromatography using Strep-Tactin Superflow Plus resin (QIAGEN). Purified protein was eluted using 50 mM NaH2PO4, 300 mM NaCl and 2.5 mM desthiobiotin at pH 8.0 and analysed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were aliquoted, flash-frozen in liquid nitrogen and stored at −80 °C. Protein concentrations were determined by NanoDrop using an extinction coefficient of 69,348 M−1 cm−1 for VEnT1.0 and 61,958 M−1 cm−1 for SpEnT1.0 and variants. Extinction coefficients for variants containing mTX were deduced through a comparison of DA_20_00 and VEnT1.0 using a BCA Protein Assay kit (Thermo Fisher). Protein yields of VEnT1.3 and SpEnT1.3 were 25 mg l−1 and 8 mg l−1 of culture, respectively.
Mass spectrometryPurified protein samples were desalted on 10,000 molecular weight cut-off Vivaspin centrifugal concentrators (Sartorius) using 0.1% acetic acid and diluted to a final concentration of 0.4 mg ml−1. Mass spectrometry was performed on a 1200 Series Agilent LC in conjunction with a Agilent 6510 QTOF. A 5-µl sample injection was performed, followed by a 1-min 5% acetonitrile (with 0.1% formic acid) isocratic wash. Protein was eluted over 1 min using 95% acetonitrile with 5% water. The resulting multiply charged spectrum was deconvoluted using Agilent MassHunter Software. Protein mass spectrometry results are shown in Supplementary Table 11.
Library constructionRounds 1, 2 and 3 for both VEnT1.0 and SpEnT1.0: site saturation mutagenesisPositions were individually randomized using degenerate NNK codons for rounds 1–3 for VEnT1.0 and rounds 2 and 3 for SpEnT1.0. For SpEnT1.0 round 1, individual mutants were prepared with the relevant mutation installed (Supplementary Fig. 15). DNA libraries were constructed by overlap extension PCR. Primers for library generation are given in Supplementary Table 12. Assembled genes and pET-29b(+) vector were digested using NdeI and XhoI endonucleases, gel-purified and subsequently ligated using T4 DNA ligase in a 5:1 ratio, respectively. Ligations were transformed into E. coli DH10b cells, the resulting colonies were pooled together, and plasmid DNA was extracted using a Miniprep Kit (QIAGEN) to yield plasmid DNA for each library. Sequencing was performed by Source BioScience.
Shuffling by overlap extension PCRAfter each round of evolution, beneficial mutations were combined by DNA shuffling of fragments generated by overlap extension PCR. Primers were designed that encoded either the parent amino acid or the identified mutation. These primers were used to generate short fragments that were gel-purified and mixed for assembly of the full-length gene by overlap extension PCR. Final full-length genes contain all possible combinations of mutations at specified positions. Genes were cloned as described above.
Library screeningFor protein expression and screening, all transfer and aliquoting steps were performed using Hamilton liquid-handling robots. Chemically competent E. coli BL21(DE3) cells harbouring pEVOL_mTX-RS/tRNACUA were transformed with the appropriate library plasmids. Freshly transformed colonies were used to inoculate 150 μl of 2× YT medium supplemented with 50 μg ml−1 kanamycin and 25 μg ml−1 chloramphenicol in Corning Costar 96-well microtitre round-bottom plates. Each plate contained six freshly transformed clones of the parent template and two clones containing pET-29b(+)_RFP as internal references. For round 1 of VEnT1.0 and rounds 2 and 3 for SpEnT1.0, one 96-well plate was assessed per library. For rounds 2 and 3 for VEnT1.1 and VEnT1.2, libraries were pooled together in groups of three, and two plates were picked from each pooled transformation. Plates were incubated overnight at 30 °C, 80% humidity in a shaking incubator at 900 rpm. Then, 40 μl of overnight culture was used to inoculate 960 μl 2× YT medium supplemented with 50 μg ml−1 kanamycin, 25 μg ml−1 chloramphenicol and 0.5 mM mTX, which was added as a 0.5-M stock dissolved in 1 M NaOH. The cultures were incubated at 30 °C, 80% humidity and 900 rpm until an OD600 of about 0.6 a.u. Protein expression was induced by the addition of l-arabinose to a final concentration of 0.05% and IPTG to a final concentration of 0.1 mM. Induced plates were incubated for 20 h at 30 °C, 80% humidity and 900 rpm. Cells were gathered by centrifugation at 2,900g for 5 min. The supernatant was discarded and the pelleted cells were resuspended in 400 μl of PBS lysis buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4, 1.0 mg ml−1 lysozyme, 0.5 mg ml−1 polymyxin B and 1 μg ml−1 DNase I) and incubated for 2 h at 30 °C, 80% humidity, with shaking at 900 rpm. Cell debris was removed by centrifugation at 2,900g for 5 min.
Rounds 1, 2 and 3 for substrate 1A 75-µl volume of clarified lysate was transferred to 96-well polypropylene microtitre plates containing 25 µl of 1.2 mM substrate 1 in PBS buffer pH 7.4 with 5% dimethyl sulfoxide (DMSO) as a cosolvent. Samples were irradiated at 405 nm in a UV curing LED oven (equipped with 365-nm and 395-nm LEDs, UV intensity 750 mW cm−2, LED module size 100 × 100, NovaChem, 100% light intensity corresponds to a photon flux density of 10,960 µmol m2 s−1), with pulsing irradiation (10 s on, 10 s off) to avoid heating and maintain the reaction temperature at 4 °C for a total irradiation time of 1 min for round 1, and 0.5 min for rounds 2 and 3 at 100% intensity. Reactions were quenched with the addition of 100 µl of acetonitrile, the plates heat-sealed and incubated for a further 1 h at 30 °C, 80% humidity and 900 rpm. Precipitated proteins were removed by centrifugation at 2,900g for 10 min. A 100-µl volume of the clarified reaction mixture was transferred to 96-well polypropylene microtitre plates and heat-sealed with pierceable foil. Reactions were evaluated by ultra-performance liquid chromatography (UPLC) analysis.
Round 1 substrate 2Individual mutants were prepared as stated in ‘Protein production and purification’ The mutants were then evaluated as described in ‘General procedure for analytical-scale biotransformations’.
Rounds 2 and 3 for substrate 2A 75-µl volume of clarified lysate was transferred to 96-well polypropylene microtitre plates containing 25 µl of 0.8 mM substrate 2 in PBS buffer pH 7.4 with 5% DMSO as a cosolvent. Samples were irradiated at 405 nm in a UV curing LED oven (equipped with 365-nm and 395-nm LEDs, UV intensity 750 mW cm−2, LED module size 100 × 100, NovaChem), with pulsing irradiation (10 s on, 10 s off) at 4 °C for a total irradiation time of 1 min for round 2, and 0.5 min for round 3 at 100% intensity. Reactions were quenched with the addition of 100 µl of acetonitrile, the plates heat-sealed and incubated for a further 1 h at 30 °C, 80% humidity and 900 rpm. Precipitated proteins were removed by centrifugation at 2900g for 10 min. A 100-µl volume of the clarified reaction mixture was transferred to 96-well polypropylene microtitre plates and heat-sealed with pierceable foil. Reactions were evaluated by UPLC analysis.
General procedure for analytical-scale biotransformationsAnalytical-scale biotransformationsBiotransformations were performed in 96-well microtitre round-bottom polypropylene plates, using HD Clear high-performance tape (Duck), 3-inch × 54.6-yard roll, to seal the samples. Biotransformations were performed at 4 °C using either 0.4 mM substrate 1 or 0.2 mM substrate 2 and the relevant biocatalyst (0.5 µM) in PBS buffer pH 7.4 with 10% DMSO as a cosolvent. Samples were irradiated at 405 nm in a UV curing LED oven (NovaChem), 23 cm below the LED array (unless stated otherwise). Instrument settings: 100% intensity, 750 mW cm−2, 10 s on/off pulse. Conditions for substrate scope characterization are detailed in Supplementary Table 7. Reactions were evaluated by UPLC analysis (see Supplementary Table 9 for methods).
Anaerobic biotransformationsFor anaerobic biotransformations with VEnT1.3, samples of enzyme and substrate 1 were incubated in a glovebox overnight on ice to ensure complete removal of oxygen. Reactions were set up in the glovebox in glass vials (final volume, 500 µl) using 0.125 mol% (0.5 µM) enzyme in PBS (pH 7.4) with substrate 1 (400 µM) and 10% DMSO as a cosolvent. Then, 25-µl samples were taken at 20 40, 60, 120, 180 and 210 s, and quenched with 1 vol of MeCN. Reactions were evaluated by UPLC analysis. For anaerobic biotransformations with SpEnT1.3, samples of enzyme and substrate 2 were incubated in a glovebox overnight on ice to ensure complete removal of oxygen. Reactions were set up in the glovebox in glass vials (final volume, 500 µl) using 1 mol% (2 µM) enzyme in PBS (pH 7.4) with substrate 2 (200 µM) and 10% DMSO as a cosolvent. Then, 25-µl samples were taken at 20 40, 80, 100 and 120 s, and quenched with 1 vol of MeCN. Reactions were evaluated by UPLC analysis (Supplementary Table 9).
Anaerobic reactions with small-molecule thioxanthoneFor anaerobic reactions, individual samples of thioxanthone, substrate 1 and substrate 2 in DMSO were incubated in a glovebox overnight on ice to ensure complete removal of oxygen. For reactions with substrate 1, reactions were set up in the glovebox in glass vials (final volume, 500 µl) using 10 mol% (40 µM) thioxanthone in PBS (pH 7.4), substrate 1 (400 µM) and 10% DMSO as a cosolvent. Then, 25-µl samples were taken at 1, 2, 3, 4, 5, 7.5, 10 and 15 min, and quenched with 1 vol of MeCN. Reactions were evaluated by UPLC analysis. For reactions with substrate 2, reactions were set up in the glovebox in glass vials (final volume, 500 µl) using 20 mol% (40 µM) thioxanthone in PBS (pH 7.4), substrate 2 (200 µM) and 10% DMSO as a cosolvent. Then, 25-µl samples were taken at 5, 10, 15, 20, 30, 40, 50 and 60 min, and quenched with 1 vol of MeCN. Reactions were evaluated by UPLC analysis (Supplementary Table 9).
Total turnover numbersTotal turnover numbers achieved by VEnT1.3 were determined as follows. VEnT1.3 (0.25, 0.1, 0.05 and 0.01 mol%)-catalysed biotransformations were performed in glass vials using 1 (400 µM) in PBS (pH 7.4) with 10% DMSO cosolvent in a 1-ml volume. Reactions were performed under general conditions and samples were taken at 1, 2, 3, 4, 5, 6, 11, 18, 23, 28, 33 and 38 min by sampling 50 µl of the reaction and quenching with 1 vol of MeCN. Reactions were evaluated by UPLC analysis. Preirradiation of VEnT1.3 for 90 min of 405-nm light led to enzyme deactivation. Total turnover numbers achieved by SpEnT1.3 were determined as follows. SpEnT1.3 (0.25, 0.1, 0.05 and 0.01 mol%)-catalysed biotransformations were performed in glass vials using 2 (200 µM) in PBS (pH 7.4) with 10% DMSO cosolvent in a 1-m volume. Reactions were performed under general conditions and samples were taken at 1, 2, 3, 4, 5, 6, 11, 18, 23, 28, 33 and 38 min by sampling 50 µl of the reaction and quenching with 1 vol of MeCN. Reactions were evaluated by UPLC analysis (Supplementary Table 9).
VEnT1.3 and SpEnT1.3 temperature profileBiotransformations were performed at 4 °C and room temperature in glass vials using 1 µM of the relevant catalyst (VEnT1.3 or SpEnT1.3) and relevant substrate (400 µM of substrate 1 for VEnT1.3 and 200 µM substrate 2 for SpEnT1.3) in PBS buffer with 10% DMSO as a cosolvent (final volume, 500 µl). For reactions at 4 °C, all reaction components were incubated in a cold room maintained at 4 °C for 30 min before running reactions. For both temperatures, reactions were run using 100% intensity irradiation at 405 nm with 10 s on/10 s off intervals and 25-µl samples were taken at 20, 40, 60, 80, 120, 150, 180, 210, 240, 260, 280, 300 s. Reactions were evaluated by UPLC analysis (Supplementary Table 9).
VEnT1.3 and SpEnT1.3 substrate concentration rate profileTo investigate the effect of substrate concentration on the rate of reaction with VEnT1.3, biotransformations were performed using 0.1 µM VEnT1.3 and a range of substrate concentrations (0, 10, 20, 35, 50, 75, 100, 125, 150, 200 and 250 µM) in PBS buffer (pH 7.4) with 10% DMSO as a cosolvent. Reactions were run in a 96-well plate with a volume of 250 µl and irradiated at 4 °C (10 s on/off pulse at 405 nm). Time points were taken at 5, 10, 15, 20, 25 and 30 s of irradiation. Then, 50-µl samples of the reaction were taken at each time point and quenched with 1 vol of MeCN. Samples were analysed by UPLC analysis. The initial rate v0 at each substrate concentration was calculated using the slope of the time course. To investigate the effect of substrate concentration on the rate of reaction with SpEnT1.3, biotransformations were performed using 0.4 µM SpEnT1.3 and a range of substrate concentrations (0, 10, 20, 30, 50, 75, 100, 125, 150, 175, 200, 225 and 250 µM) in PBS buffer (pH 7.4) with 10% DMSO as a cosolvent. Reactions were run in a 96-well plate with a volume of 250 µl and irradiated at 4 °C (10 s on/off pulse at 405 nm). Time points were taken at 1, 2, 3, 4, 5, 6 s of irradiation for 10, 20, 35 and 50 µM and at 5, 10, 15, 20, 25 and 30 s of irradiation for 75, 100, 125, 150, 200 and 250 µM. Then, 50-µl samples of the reaction were taken at each time point and quenched with 1 vol of MeCN. Samples were analysed by UPLC analysis (Supplementary Table 9). The initial rate v0 at each substrate concentration was calculated using the slope of the time course.
VEnT1.3 and SpEnT1.3 light intensity rate profileTo investigate the effect of light intensity on the rate of reaction of VEnT1.3 with substrate 1, biotransformations were performed in 2-ml MS glass vials using 1 µM VEnT1.3 and 400 µM 1 in PBS buffer (pH 7.4) with 10% DMSO as a cosolvent (total reaction volume, 500 µl). Reactions were positioned 3 cm from the LED array and irradiated as described at varying LED intensities and time points taken at 20, 40, 60, 80, 100 and 120 s for 10% and 20%, and at 10, 20, 30, 40, 50 and 60 s for 30%, 40%, 50% and 100% and analysed by UPLC analysis. The initial rate v0 at each substrate concentration was calculated in triplicate using the slope of the time course at less than 10% conversion. To investigate the effect of light intensity on the rate of reaction of SpEnT1.3 with substrate 2, biotransformations were performed in 2-ml MS glass vials using 1 µM SpEnT1.3 and 200 µM 2 in PBS buffer (pH 7.4) with 10% DMSO as a cosolvent (total reaction volume, 500 µl). Reactions were positioned 3 cm from the LED array and irradiated as described at varying LED intensities and time points taken at 20, 40, 60, 80, 100 and 120 s for 10% and 20%, and at 10, 20, 30, 40, 50 and 60 s for 30%, 40%, 50% and 100% and analysed by UPLC analysis (Supplementary Table 9). The initial rate v0 at each substrate concentration was calculated in triplicate using the slope of the time course at <10% conversion.
Semipreparative-scale biotransformation of 1Substrate 1 (12 mg) was dissolved to a concentration of 400 µM in PBS buffer (pH 7.4) with 10% DMSO as a cosolvent (reaction volume, 140 ml) with 2 µM VEnT1.3 (0.5 mol%). The solution was irradiated as described at 405 nm for a total reaction time of 10 min in a Pyrex dish (22 cm diameter; solution path length, 0.37 cm). Once full conversion was reached (as monitored by UPLC), the solution was transferred to a separatory funnel and extracted with 3 × 20 ml ethyl acetate and the combined organic layers were washed with 3 × 20 ml brine, dried over MgSO4, filtered and concentrated in vacuo to afford optically pure 1a (99% e.e., 11.6 mg, 97% turn yield), which required no further purification.
Semipreparative-scale biotransformation of 2Substrate 2 (10 mg) was dissolved to a concentration of 200 µM in PBS buffer (pH 7.4) with 5% DMSO as a cosolvent (reaction volume, 136 ml) with 6 µM SpEnT1.3 (3 mol%). The solution was irradiated as described at 405 nm for a total reaction time of 5 min in a Pyrex dish (22 cm diameter; solution path length, 0.37 cm). Once full conversion was reached (as monitored by UPLC), the solution was transferred to a separatory funnel and extracted with 3 × 20 ml ethyl acetate and the combined organic layers were washed with 3 × 20 ml brine, dried over MgSO4, filtered and concentrated in vacuo to afford 2a which was further purified through flash column chromatography to yield 2a (99% e.e., 16:1 d.r., 8 mg, 80% isolated yield).
Chromatographic analysisFor UPLC analysis, reactions were quenched at the stated time points with the addition of 1 vol of MeCN. Samples were shaken at 900 rpm for 1 h and precipitated proteins were removed by centrifugation (2,900g for 10 min). For chiral HPLC analysis, the substrates and products were transferred into 1.5-ml microcentrifuge tubes and extracted with 3 vols of ethyl acetate. Precipitated proteins were removed by centrifugation (14,000g for 15 min), and the organic phase was separated and directly injected onto the UPLC.
UPLC analysis was performed on a 1290 Infinity II LC system (Agilent) with a Kinetex 5-µm XB-C18 100-Å LC column, 50 × 2.1 mm (Phenomenex). Peaks were assigned by comparison with chemically synthesized standards and the peak areas were integrated using Agilent’s OpenLab software. The reverse-phase separation methods for all substrate(s)/product(s) and extinction coefficients used to calculate the yields are reported in Supplementary Table 9. Chiral analysis was performed using a UPLC 1290 system (Agilent). Enantiomers of all reaction products 1a, 2a, 5a–9a were separated using a Daicel 14S84 CHIRALPAK OD-3 column, 3 mm × 50 mm × 3 µm. For all adducts (1a, 2a, 5a–9a), the major stereoisomer formed in the biotransformations was assigned on the basis of an analogy to the SpEnT1.3-derived (S,S)-2a. Peaks were assigned by comparison with chemically synthesized standards (Supplementary Information, ‘Chemical procedures’) and peak areas were integrated using Agilent’s OpenLab software. Chiral separation methods for all substrate(s)/product(s) enantiomers used to determine the e.e. are reported in Supplementary Table 10.
Laser photoexcitation measurementsLaser photoexcitation experiments were carried out at 4 °C using an Edinburgh Instruments LP980 transient absorption spectrometer. Samples contained 100 µM small-molecule thioxanthone or VEnT/ SpEnT enzymes in PBS buffer (pH 7.4) and O2 was removed where necessary by incubation in an anaerobic glovebox (Belle Technology) for 4 h. Triplet formation was initiated by excitation at 355 nm (~50 mJ), using the third harmonic of a Q-switched Nd-YAG laser (NT342B, EKSPLA) in a cuvette of 1-cm path length. Time-dependent absorbance difference spectra were recorded with an intensified charge-coupled device detector (Andor Technologies) using a gate width of 100 ns and five averages. Time-gated fluorescence emission spectra were recorded over 100 ns between 300 nm and 700 nm using five averages. Kinetic transients were recorded using single laser pulses at the specified wavelengths with the detection system (comprising probe light, sample, monochromator and photomultiplier tube detector) at right angles to the incident laser beam. Time constants were observed from the average of at least five time-dependent absorption measurements by fitting them to a double-exponential function using L900 software (Edinburgh Instruments).
Preparation of chiral standardsA 500-µl analytical-scale biotransformation of SpEnT1.3 (3 mol%) with substrate 2 (200 μM) was performed and the resulting material extracted in ethyl acetate; then the solvent was removed in vacuo. The absolute stereochemistry was determined by vibrational circular dichroism (VCD) spectroscopy as outlined below.
Absolute configuration determination by VCD spectroscopyExperimental detailsInfrared (IR) and VCD spectra were recorded on a Bruker Vertex 70/PMA 50 VCD spectrometer at 4 cm−1 spectral resolution by accumulating 32 scans for the IR and ~40,000 scans (9 h accumulation time) for the VCD. The samples were dissolved in CDCl3 at the concentration given in the respective captions and measured using a BaF2 IR cell with an optical path length of 100 μm. Baseline correction of the VCD spectra was done by subtraction of the spectra of the solvent recorded under identical conditions.
Computational detailsDeriving the absolute configuration from the experimental spectra requires the computation of IR and VCD spectra. Therefore, a conformational sampling was carried out based on a systematic search algorithm at the force-field level (MMFF)52,53. All so-obtained conformers were subjected to further geometry optimizations at B3LYP/def2tzvp/IEFPCM(CHCl3) level of theory using Gaussian 09 rev E.0154. For the final comparison with the experiment, the IR and VCD spectra were simulated from the single-conformer spectra using the ΔEZPC‐based Boltzmann weights and by assigning a Lorentzian band shape with half‐width at half‐height of 6 cm−1 to the computed dipole and rotational strength.
Analysis of the spectraAs the relative configuration of (S,S)-2a was known, only the absolute configuration had to be determined by VCD spectroscopy. Only two conformers were found in the conformational analysis: the lowest-energy conformer is shown in Supplementary Fig. 26, while the second conformer differs in the relative orientation of the benzyl group and possesses an energy difference of 1.3 kcal mol−1. The comparison of the experimental and computed spectra unambiguously confirms the (S,S)-configuration.
Crystallization, refinement and model buildingVEnT1.0, VEnT1.3 and SpEnT1.3 were crystallized by mixing 200 nl of 15 mg ml−1 protein in 20 mM HEPES buffer, pH 7.5, with equal volumes of precipitant. All trials were conducted by sitting-drop vapour diffusion and incubated at 20 °C. Crystallization conditions were identified using the PACT and SG1 screens (Molecular Dimensions) and are as follows. VEnT1.0: 0.1 M MIB, pH 7.5, 25% PEG 1500; VEnT1.3: 0.02 M sodium/potassium phosphate, 20% PEG 3350; SpEnT1.3: 0.1 M sodium HEPES, pH 7.5, 25% PEG 3350.
Before data collection, crystals were cryoprotected by the addition of 20% PEG 200 to the mother liquor and plunge-cooled in liquid nitrogen. All data were collected at Diamond Light Source (Harwell, UK) using beamline i03. Data reduction was performed with DIALS and the structure solved by molecular replacement using a search model derived from the structure of EnT1.3 (PDB: 7ZP6). Iterative rounds of model building and refinement were performed in Coot and phenix.refine55, respectively. Validation with MolProbity and PDB-REDO56 were incorporated into the iterative rebuild and refinement process. Data collection and refinement statistics are shown in Supplementary Table 4. The coordinates and structure factors for VEnT1.0, VEnT1.3 and a SpEnT1.3 have been deposited in the PDB under accession numbers 9FYU, 9FYV and 9G65, respectively. Unrestrained molecular docking of the ligands was performed using Molsoft ICM-Pro.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Comments (0)