
Remember me
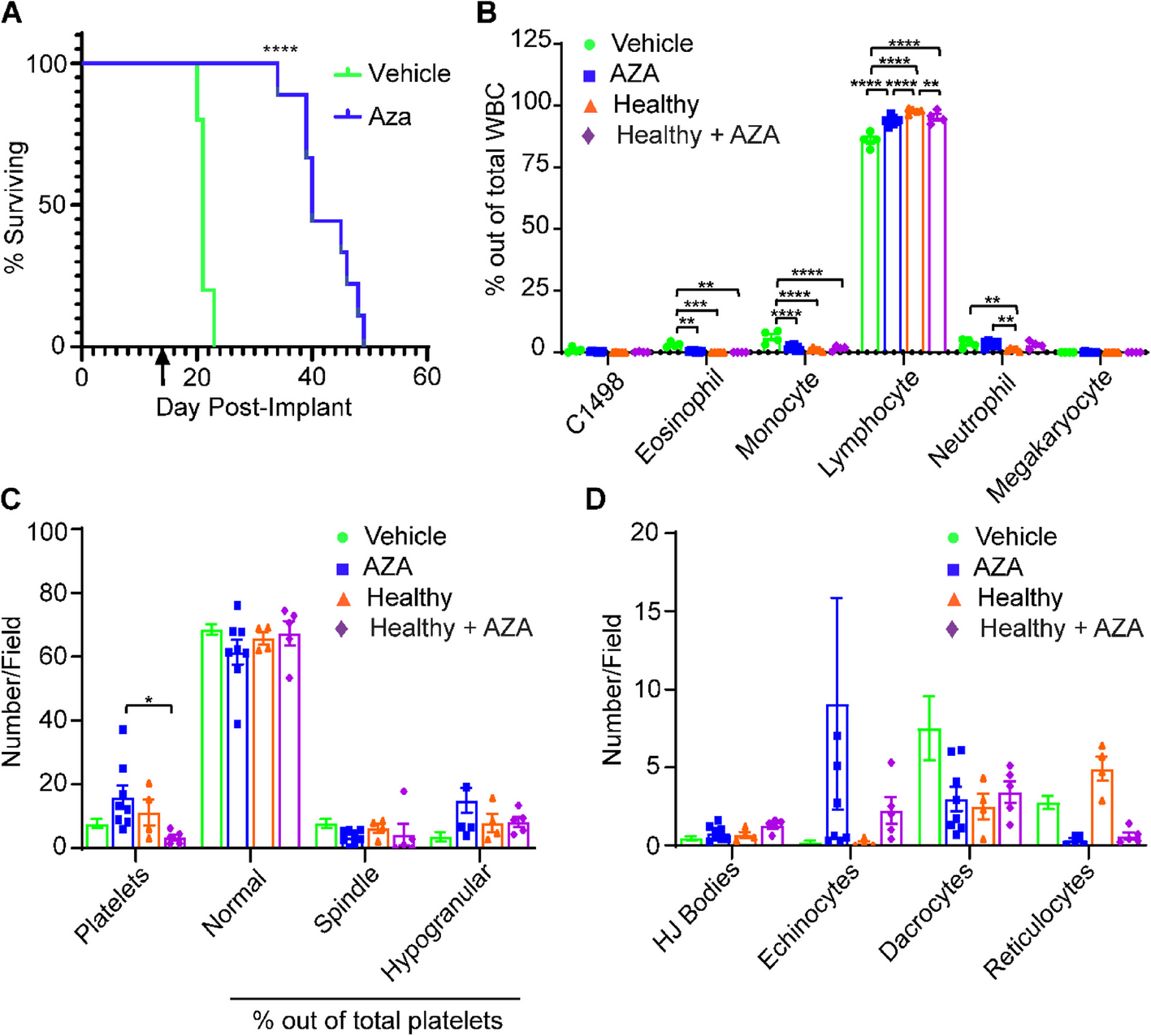





Mental stress can result in depigmentation or inhibition of hair growth (Zhang et al. 2024). We therefore generated a CUMS mouse model to observe melanin loss. A flow chart of the study is shown in Fig. 1A. Body weight reflects the stress state of mice. As shown in Fig. 1A, the body weight of mice in the control group increased steadily over time, while it was lower in the CUMS group (p < 0.001). Behavioral tests can reflect a stressed state. In the open field test, CUMS mice displayed significantly reduced total distance traveled (p < 0.001), speed of movement (p < 0.001), and time spent in the center zone (p < 0.05) (Fig. 1B), indicating a significant reduction in spontaneous activity. Similarly, CUMS significantly increased the total immobility time in the forced swimming test and the tail suspension test (p < 0.05) (Fig. 1B), indicating that CUMS can lead to depression-like behaviour in mice.
Fig. 1
Optimization of Chronic Unpredictable Mild Stress (CUMS) induced hair and skin whitening mouse model. A Timeline of CUMS model and body weight change. B Behavioral test of CUMS, including FST, TST, SPT, and OFT with trajectory record. C Serum IFN-γ, IL-6, and CXCL10 detected by ELISA kit. D Full-length portraits taken by Canon D30 revealed that CUMS induced dorsal hair and tail skin to turn white E. Dermatoscope images were captured by device 3Gen DermLite DL4, F wood’s light photos of Control and CUMS group. G Melanin in epidermis and follicles of mouse tail skin. (Left are from control group, and right from CUMS. Fontana-Masson, HE stained × 400, scale bar = 50 μm. Data are from three independent experiments). Each sample is represented as one dot. P-values were calculated using two-sided Mann–Whitney U-test. *p < 0.05, ** p < 0.005 and *** p < 0.001. Abbreviations: FST, Forced Swimming Test; TST, Tail Suspension Test; SPT, Sucrose Preference Test; OFT, Open Field Test
To evaluate the impact of CUMS on melanin synthesis, we photographed mice using a Canon camera and a dermatoscope with Wood’s lamp. Compared with control mice, CUMS mice exhibited more depigmentation in the dorsal skin and tail (Fig. 1C–E). Haematoxylin & eosin staining combined with Fontana-Masson staining revealed melanin particles in the epidermis and hair follicles of the tail skin in CUMS mice (Fig. 1F). ELISAs showed that inflammatory cytokines associated with vitiligo, such as IFNγ, IL6, and CXCL10 were significantly elevated in CUMS serum (Fig. 1G). Together, these findings indicate that CUMS impeded melanin synthesis.
Sequencing of single skin cells from control and CUMS mice reveals diversity in skin cell populationsWe conducted single-cell RNA sequencing (scRNA-seq) on mouse tail skin using the BD Rhapsody System (Fig. 2A). This generated transcription profiles of 72,011 cells. The data from multiple sequencing runs were integrated into a unified Uniform Manifold Approximation and Projection (UMAP), which identified 11 different cell clusters (Fig. 2B). These included keratinocytes (Krt15, Krt14), fibroblasts (Col1a2, Col1a1, Dcn), melanocytes (Tyr, Kit, Mitf), upper hair follicle cells (infundibulum, Krt17, Krt79, Krt6a), immune cells (Cd3, Cd68, Cd14, Cd74, H2-Eb1), endothelial cells (Pecam1, Cldn5, Vwf), sebaceous gland cells (Scd1, Mgst1), outer bulge cells (Postn, Ptn, Krt5, Krt17), inner bulge cells (Krt6a, Krt75, Krt5, Krt17, Ptn), Schwann cells (Mbp, Kcna1), and pericytes (Rgs4, Rgs5, Kcnj8). Classical marker genes were highly expressed in all 15 cell types (Fig. 2C). All cell subtypes were present in both the CUMS and control groups (Fig. 2D). A boxplot of cell proportions showed a decrease in melanocytes and an increase in immune cells in the tail skin of the CUMS group compared with the control group (Fig. 2E). A heatmap based on the ro/e index displayed a consistent distributional preference among the cells (Fig. 2F).
Fig. 2
scRNAseq identifies well-defined cell populations in CUMS mice skin. A Flow chart of single cell sequencing. B Uniform Manifold Approximation and Projection (UMAP) plot for 14,211 high quality single cell transcriptomes from mouse skin with Control (n = 3) and CUMS (n = 3), revealing 11 different cell populations. C Dot plot displaying top expressed genes in each cell population compared to the other cell populations identified in skin. D Fractions of cell populations identified in the skin of Control and CUMS. E Boxplot of the distribution of eleven selected cell populations identified in the skin of Control and CUMS. F Tissue preference of each cluster estimated by the Ro/e index, which Ro/e > 1 suggests enriched, and Ro/e < 1 suggests depleted. Abbreviations: IB, inner bulge; KC, keratinocytes; OB, outer bulge; SG, Sebaceous gland; UFC, upper hair follicle cells (infundibulum)
Impaired maturation and senescence of hair follicle melanocyte stem cells in CUMS mice skinClustering analysis defined four melanocyte sub-clusters, Mel1–Mel4 (Figure S2a). The top five marker genes heatmap for each sub-cluster revealed that the Mel1 cluster expressed keratinocyte-related markers, such as Krt14, Krt5, Krtdap, Krt1, and Krt10, which was quite different from the other three melanocytes sub-clusters (Figure S2b,c). Moreover, the GO enrichment analysis of Mel1 cells also showed Mel1 cells were enriched in cytoplasmic translation, translation at synapse, translation at pre-synapse, translation at post-synapse (Figure S2 d), which we presumed that these represented keratinocytes containing transferred melanin granules, and were therefore excluded from the melanocyte sub-clusters (Fig. 3A).
Fig. 3
Characterization of the melanocyte population in mouse skin by scRNASeq. A UMAP plot for melanocyte cluster, revealing three different cell populations. B Dot plot displaying top expressed genes in each melanocyte cluster. C. Heatmap of Top 5 marker gene expression of three melanocyte clusters. D. Boxplot of the distribution of three selected cell populations identified in melanocyte clusters. E. Ro/e index of each cluster. Ro/e > 1 suggests enriched, and Ro/e < 1 suggests depleted. F. The inferred pseudo temporal trajectories of all cells using Monocle 3. Cells were colored by the inferred pseudo time. G. Heatmap of transcription factor activity of each cluster. H. Gene Set Variation Analysis (GSVA) for cell death modes and related pathways in each melanocyte cluster. I. Violin blots of pyroptosis, Oxidative stress, and mitochondrion disfunction. Each sample is represented as one dot. P-values were calculated using two-sided Mann–Whitney U-test. *p < 0.05
Both cell proportions and the ro/e index showed a significantly decreased Mel2 subcluster in the CUMS group (Fig. 3D,E). Mel2 expressed high levels of melanocyte stem cell (McSC) markers (Dct, Mitf, Pax3, Sox10, and Kit), but lacked expression of melanin synthesis and proliferation markers (Tyr, Tyrp1, MelanA, and Pmel) (Fig. 3B,C). Consequently, we identified the Mel2 cluster as McSCs, while the Mel3 cluster represented mature melanocytes. To study the differentiation of melanocytes, we arranged the cells in pseudo-time order using monocle3 and observed a trajectory from Mel2 to Mel3 differentiation. During the differentiation process, a gradual reduction of McSC markers such as Mitf, Pax3, Sox10, and Kit was noted, further supporting the stem cell identity of Mel2 (Fig. 3F). Mel2 cells expressed high levels of genes such as Txnip, Egr1, and Fos, and also exhibited increased predicted transcription factor activity for Junb, Fos, Prrx2. Notably, Fos was enriched in Mel2 at both expression and predicted transcription factor activity levels (Fig. 3C, G). The transcription factor “Fos” is rapidly induced by stimuli such as growth factors, cytokines, and stress. “FOS family” proteins can form heterodimers with “JUN family” proteins to create the Activator Protein-1 complex, regulating gene expression of cell proliferation, differentiation, necroptosis, pyroptosis, and immune responses (Angel et al. 2001; Zhang et al. 2025). Enrichment analysis indicated that Mel2 cells were significantly enriched in processes related to pigmentation and pigment cell differentiation, while Mel3 cells were enriched in pigmentation and developmental pigmentation processes (Figure S2e,f). Heatmap of cell death showed that Mel2 cells were prone to pyroptosis and necroptosis, while Mel3 cells were more susceptible to oxidative stress, mitochondrial dysfunction, and ferroptosis (Fig. 3H,I). Immunohistochemistry confirmed the existence of the Mel2 and Mel3 clusters and also showed pyroptosis in the Mel2 cluster (Fig. 4).
Fig. 4
Immunofluorescence of Melanocytes from mouse tail skin. A Mel2 represented McSCs reduced in CUMS. DAPI (Blue), DCT (red), KRT15 (Green), CD34 (Yellow). B Mel3 represented Mature Melanocytes reduced in CUMS. DAPI (Blue), DCT (red), C-Kit (Green), MelanA (Yellow). C Pyroptosis was increased in CUMS. DAPI (Blue), DCT (red), GSDMD (Green), NLRP3 (Yellow). scale bar = 50 μm, Partial enlarged view scale bar = 20 μm. Data are from three independent experiments. Abbreviations: MC, melanocyte; McSC, melanocyte stem cell
Fig. 5
Characterization of the immune cells population in mouse skin by scRNASeq. A Immune cells t-Distributed stochastic neighbor embedding (t-SNE) plot. B Dot plot displaying top expressed genes in three immune cells clusters. C Bar plot showing the fractions of cell populations identified in mouse tail skin. D Muti-volcano plot of distribution of gene variation in CUMS and Control. E Dot plot displaying top expressed genes in three T cells clusters. F T cell function related violin plot. G Dot plot displaying top expressed genes in three T cells clusters. H Pie charts of proportions of three T cells clusters in CUMS and Control. I Violin plots of Macrophages signature. J Phagocytic function of Macrophages. K Antigen presentation scores of dendritic cells. P-values were calculated using two-sided Mann–Whitney U-test. *p < 0.05, ** p < 0.005 and *** p < 0.001. Abbreviations: DC, dendritic cells; Th1, helper T cells.; Trm, resident memory T cells
Inflammatory endogenous environment of hair follicle cell populations in the tail skin of CUMS miceThe hair follicle niche in which McSCs are located plays an important role in maintaining their differentiation ability (Huang et al. 2024). Thus, we classified hair follicle cells into four clusters: hair follicle stem cells (HFSCs), upper hair follicle cells (infundibulum, UFCs), outer bulge cells (OBs), and inner bulge cells (IBs), with each cluster annotation shown in Figure S2a–d. McSCs are located in the bulge and hair germ (HG) area and are adjacent to HFSCs. We therefore focused on the reduced number of HFSCs in the CUMS group and found a significant increase in the expression of pro-inflammatory chemokines (Il1, Il6, Il15, Ccl2, Cxcl12) (Figure S3e). Overall, the expression of genes related to the γ-interferon pathway, such as Ifngr1 and Jak1, was obviously increased in the HFSCs cluster (Figure S3f,g). These findings indicate that McSCs in the CUMS group are within an inflammatory internal environment.
Keratinocyte and fibroblast populations in CUMS mouse tail skin concentrate on pro-inflammatory responsesIn vitiligo lesions, stromal cells, especially keratinocytes and fibroblasts, are extensively involved in the inflammatory destruction of melanocytes by secreting various chemokines (Shi et al. 2024; Yang et al. 2023). The majority of cells sequenced in our scRNA-seq analysis constituted keratinocytes (42,408 cells). Sub-clustering analysis revealed significant proportional differences in the nine sub-clusters. These nine sub-clusters could be identified as four keratinocyte states based on previously reported expression profiles (Figure S4a). Hair follicle keratinocytes highly expressed Krt15 and Krt17. Super-spinous keratinocytes expressed Slurp1 and Sprr1b. Spinous keratinocytes specifically expressed early differentiation keratins (Krt1 and Krt10). Basal keratinocytes highly expressed Krt14, Mt2, Col17a1, and Dst (Figure S4b,c). A deeper analysis revealed that all keratinocyte populations in the CUMS group were focused on inflammation and immune responses, including the interferon pathway, oxidative stress responses, and cytokine signalling (Figure S4 d).
Cutaneous fibroblasts (FCs) are heterogeneous and participate in multiple skin disorders(Plikus et al. 2021; Shi et al. 2024; Yang et al. 2023), including psoriasis, atopic dermatitis, and vitiligo (Cai et al. 2023; He et al. 2020; Xu et al. 2022). We explored the heterogeneity of FCs in the CUMS group and identified four subgroups. These subgroups were further categorised into Col1a1 + FCs (Col1a1, Col3a1, CD34, Dcn), and Pdgfa + FCs (Pdgfa, Spon1, Serpine2) for second-level analysis (Figure S4e,f). The Col1a1 + FCs cluster was significantly increased in CUMS mice and highly expressed the pro-inflammatory chemokines Il1b, Il6, Mmp2 and Ccl2 (Figure S4 g,h). Thus, this cluster represents pro-inflammatory fibroblasts that regulate the recruitment and organization of lymphocytes and myeloid cells.
γδT initiates nonspecific immunity in the tail-depigmented skin region of CUMS miceNext, we attempted to characterize the dynamics of the immune microenvironment in CUMS mouse tail skin. Immune cells in mouse tail skin were categorised into three clusters: T cells (Cg3 g, Cd3e, Cxcr6), Macrophages (Cd68, Cd14), and Dendritic cells (Cd74, H2-Eb1, Cd207, Ltc4 s). Compared with the control group, a t-distributed stochastic neighbor embedding (t-SNE) plot, and bar charts showed CUMS mice to have increased proportions of T cells and macrophages among immune cells (Fig. 5 A-D). We then evaluated the function of T cells and found that processes such as chemotaxis, cytotoxicity, and extravasation were all predicted to be enhanced in the CUMS group (Fig. 5E). Further analysis of T cells revealed three sub-clusters: γδT cells (Trdc, Trgc1, Tob2), Th1 cells (Ctla2a, Cd28, Cd4), and CD8 + resident memory T cells (CD8 + Trm) (Tex14, Cd52) (Fig. 5F,G). Dramatic increases in the numbers and proportion of γδT cells and Th1 cells were observed in CUMS mice compared with control mice, while numbers of CD8 + Trm remained unchanged, and only a decrease in its relative proportion due to the increase in γδT in the CUMS group (Fig. 5H).
Macrophages in the CUMS group expressed high levels of inflammatory factor genes, such as Tnf, Ccl2, Ccl3, Ccl4, and Ccl22, and were more inclined to an M1 pro-inflammatory phenotype with enhanced phagocytic function. (Fig. 5I). Dendritic cells in the CUMS group showed enhanced migration capacity with higher expression of migratory DC (migDC) genes, such as Zbtb46, Flt3, and Ccr7. Antigen-presentation scores were also increased (Fig. 5J).
To fully understand the differentiation trajectory of T cells in the skin of CUMS mice, we used monocle analysis to reveal three major T cell pathways. Path 1 started with CD8 + Trm, and Paths 2 and 3 ended with Th1 and γδT states, respectively. CUMS caused gradual activation with the change in pseudo-time (Fig. 6A,B). We found that the cumulative γδT cell density increased gradually with pseudo-time, which was supported by the expression dynamics of γδT signature genes along the inferred pseudo-time axis (Fig. 6C,D). The pseudo-time heatmap revealed that expression of T cell proliferation genes, such as Il2ra and Il2rb, and inflammatory factor genes, such as Il17a and Tnf, gradually increased with CUMS exposure (Fig. 6E). In addition, GO and KEGG enrichment analyses showed that γδT cells were enriched in immune response activation, such as activation of NF-KB, TNF and JAK-STAT pathways (Fig. 6F). Together, these results indicate activation of innate immunity in depigmented skin and potential initiation of specific immunity characterised by enhanced migratory capacity of antigen-presenting cells.
Fig. 6
Characterization of the γδT cells population in mouse skin. A-B The inferred pseudo temporal trajectories of all T cells in skin using Monocle 3. Cells were colored by the inferred pseudo time. C Genes expression dynamics of Trdc, Trgc1, Il17a and Tnf of different T cells clusters, along the inferred pseudo-time axis. D Cumulative-density plot of different clusters of T cells. E The pseudo-time heat map of T cell proliferation genes and inflammatory genes during the occurrence of CUMS. F GO and KEGG enrichment analysis of γδT cells. Abbreviations: Th1, helper T cells; Trm, resident memory T cells
Ligand-receptor analysis reveals cell type-specific networks in CUMS mouse skinWe employed the CellChat package to elucidate the complex relationship between different clusters. By comparing the overall communication probability between the control and CUMS groups, we identified 33 enriched signalling pathways, with eight pathways being more prevalent in the CUMS group, including CXCL, NRG, NPR2, CD137, NT, GDF, OSM, and CCL (Fig. 7A). These pathways may therefore contribute to the progression of vitiligo. The CXCL and CCL signaling pathways in CUMS mice ranked highly in network visual chord diagrams (Fig. 7B). CXCL16-CXCR6 is the most significant ligand-receptor pair in CXCL signalling (Fig. 7C); therefore, we further examined the expression of these chemokines in all cell clusters at the mRNA level (Fig. 7D). Dendritic cells expressed significantly higher amounts of Cxcl16, and T cells, especially γδT cells, expressed Cxcr6 according to the gene distribution UMAP (Fig. 7E). Cxcl16-positive DCs expressed more genes related to differentiation and mutation, like Cd83, Cd80, CD86, and Cxcr6-positive γδT cells expressed more pro-inflammatory cytokines such as Il17, Il2, Tnf (Fig. 7F,G). These results indicate that the CXCL16-CXCR6 ligand-receptor may participate in the immune infiltration. Detailed Cellchat information of the different cells with the Mel2 and Mel3 clusters is presented in Figure S5.
Fig. 7
Cellchat of the immune cells with other cells from the mouse skin. A Significant signaling pathways were ranked based on differences in the overall information flow within the inferred networks between CUMS and Control skin scRNA-seq dataset. B Network visual chord diagram showing the CXCL and CCL signaling pathways ranked high in CUMS. C Comparison of the multiple ligand–receptor pairs among different cells. D Violin plots displaying the vary expression of CXCL16 and CXCR6 in all cell clusters at the mRNA level. E The feature plots showing CXCL16 and CXCR6 mRNA expression within total skin cells between CUMS and Control. F Dot plot showing different cytokines in each T cell cluster of skin. G Dot plot showing cytokines of Cxcl16-negative DC and Cxcl16-positive DC; cytokines of Cxcr6-negative γδT and Cxcr6-positive γδT. P-values were computed from one-sided permutation test (CellChat 1.5.0). *p < 0.05, ** p < 0.005 and *** p < 0.001. Abbreviations: DC, dendritic cells; HFSC, hair follicle stem cell; IB, inner bulge; KC, keratinocytes; OB, outer bulge; SG, Sebaceous gland; Th1, helper T cells; Trm, resident memory T cells; UFC, upper hair follicle cells (infundibulum)
Subsequent activation of nonspecific and specific immunity in the spleen of CUMS miceTo investigate whether the migrating dendritic cells in the skin of CUMS mice affect the immune function of the spleen, we performed scRNA-seq on the spleen. After quality control, scRNA-seq data from mouse spleen were classified into five clusters: B cells (Cd19, Ms4a1, Cd79a), T/Natural killer (NK) cells (Cd3, Trbc2), Plasma cells (Igkc, Iglc1, Jchain), Myeloid cells (Csf1r, Cd68, Cd14), and Proliferating cells (Top2a, Mki67, Pclaf) (Fig. 8A,B). A notable increase in the proportion of T/NK cells was identified in CUMS spleen compared with control spleen (Fig. 8C). Subsequently, secondary analysis isolated seven sub-groups: CD4 + naive T cells (Lef1, Tcf7, Satb1), CD4 + effector memory T cells (Egr3, Egr2, Nr4a3), IFN-γ + T cells (Ifit3b, Ifit1, Ifit3), regulatory T cells (Foxp3, Il2ra, Ctla4), CD8 + cytotoxic T cells (Cd8b1, Cd8a, Ccr9), CD8 + effector memory T cells (Myb, Eomes, Slamf7), and NKT cells (Id2, Cxcr6, Klrb1c, Ccr9) (Fig. 8D,E). An increase in the proportion of CD8 + effector memory T cells, regulatory T cells, and NKT cells was identified (boxplot and Ro/e index, Fig. 8F,G). NKT cells expressed the highest levels of most pro-inflammatory cytokines, like Ifng (Fig. 8G). Therefore, we evaluated the functions of T cells, particularly NKT cells, which were predicted to be enhanced to varying degrees for chemotaxis, cytotoxicity, and extravasation (Fig. 8K).
Fig. 8
Characterization of the immune cells population in mouse spleen by scRNASeq. A UMAP of immune cells in mouse spleen. B Dot plot displaying top expressed genes in five immune cells clusters. C Bar plot showing the fractions of cell populations identified in mouse spleen. D UMAP of T cells clusters. E Dot plot displaying top expressed genes in seven T cells clusters. F Ro/e index of each cluster. Ro/e > 1 suggests enriched, and Ro/e < 1 suggests depleted. G Dot plot showing different cytokines and receptors in each T cell cluster. H Violin plots of Macrophages signature. I Comparison of the multiple ligand–receptor pairs among NKT with other cells. J Violin plots displaying the vary expression of CXCL16 and CXCR6 in all T cell clusters at the mRNA level in CUMS and Control. K NKT cell function related violin plot. P-values were computed from one-sided permutation test (CellChat 1.5.0). *p < 0.05, Abbreviations: DC, dendritic cells; Mac, macrophages; NKT, natural killer T cells; Tem, effector memory T cells; Tcyt, cytotoxic T cells; Treg, regulatory T cells
To further explore whether NKT cells in the spleen of CUMS mice are activated by antigen-presenting cells, we conducted CellChat package analysis. We found Ccl5-Ccr5 and Cxcl16-Cxcr6 to be the most significant two pairs between cDC2 and NKT (Fig. 8I). Gene expression analysis showed that dendritic cells, more specifically the cDC2 cluster, expressed the highest level of Cxcl16. NKT cells expressed the highest level of Cxcr6 in the mouse spleen (Fig. 8J). Immunohistochemistry confirmed the expression of Cxcl16 in cDC2 and Cxcr6 in NKT cells within the CUMS mouse spleen (Fig S6). Together, these results indicate the initiation of specific and non-specific immune responses represented by CD8 + Tem and NKT by the spleen after recognition of antigens and migration in cutaneous DCs.
Highly cytotoxic γδT cells are also present in the marginal zones of vitiligo lesionsCD8 + T cell infiltration is a pathological feature of vitiligo lesions. However, previous studies have rarely reported the presence of γδT cells in the marginal zones of vitiligo lesions. Therefore, to search for evidence of the presence of γδT cells in skin lesions from patients with vitiligo, we downloaded a scRNA-seq dataset from the Genome Sequence Archive (accession number PRJCA006797), which includes normal skin samples from five healthy controls and marginal zone skin samples from 10 vitiligo patients.
After quality control, transcriptomes of 48,887 cells were retrieved (Fig. 9A). An increased proportion of T cells was observed in vitiligo samples compared with control samples (Fig. 9B). The T cell clusters were extracted and further unbiased clustering identified six T cell sub-clusters: CD4 + naive T cells, Th17, CD4 + Tregs, CD8 + cytotoxic T cells (CD8 + Tcyt), CD8 + resident memory T cells (CD8 + Trm), and γδT cells. Consistent with our scRNA-seq study in CUMS mice, γδT cell numbers were increased in the marginal zones of vitiligo patients, although they accounted for a smaller proportion (Fig. 9C,D). Interestingly, CD8 + Trm cells also expressed γδT markers, indicating that CD8 + Trm cells may have acquired some functional characteristics of γδT cells, such as an early rapid immune response (Fig. 9E).
Fig. 9
Characterization of the immune cells population in human skin by scRNASeq from dataset of GEA. A UMAP plot for 48,887 high quality single cell transcriptomes from dataset of GEA of human skin with Control (n = 5) and vitiligo (n = 10), revealing eight different cell populations. B Fractions of cell populations identified in the skin of Control and vitiligo. C UMAP of T cells clusters. D Dot plot displaying top expressed genes in each cell population compared to the other cell populations identified in skin. E Violin plot showing expression of γδT markers in six T cells clusters. F Volcano plot displaying the differential genes of γδT cells. G The inferred pseudo temporal trajectories of all T cells using Monocle 3. Cells were colored by the inferred pseudo time. H Dot plot of cytotoxicity function of all T cells. I Violin plots of chemotaxis and cytotoxicity function of all T cells. J. Violin plots displaying the vary expression of CXCL16 and CXCR6 in all T cell clusters at the mRNA level in healthy and vitiligo. P-values were calculated using two-sided Mann–Whitney U-test. *p < 0.05, ** p < 0.005 and *** p < 0.001. Abbreviations: Th, helper T cells.; Trm, resident memory T cells; Tcyt, cytotoxic T cells; Treg, regulatory T cells
Differential analysis indicated that γδT cells expressed high levels of certain genes, such as MYOM2, FGR, and CX3 CR1 (Fig. 9F). In addition, γδT cells expressed high levels of T cell cytotoxic genes, such as GZMA, GZMB, and NKG7, and showed the highest cytotoxicity scores calculated by AUCell (version 1.22.0) (Fig. 9H,I). Pseudotime analysis revealed that γδT cells did not originate from other CD4 + or CD8 + T cells, displaying a distinct developmental state (Fig. 9G). Lastly, CXCL16-CXCR6, which plays an important role in cell communication in CUMS mice, was widely expressed across T cell sub-clusters and showed inter-group differences in γδT and CD8 + Trm cells (Figs. 9J and 10).
Fig. 10
Hypothesis schematics of comprehensive immune microenvironment in an optimized CUMS-induced leukoderma and leukotrichia mouse model. Left is pigmented skin and hair in the control group, and the right is depigmented skin with leukotrichia in CUMS mice. CXCR6 + γδT cells, recruited by CXCL16, produce IFNγ that induce the secretion of CXCL9/10 by keratinocytes and fibroblasts, contributing to a pro-inflammatory microenvironment, resulting in direct or indirect injury of melanocytes in epidermis and follicles (Mel3) and McSCs (Mel2). Impaired melanocytes can release melanocyte-specific antigens and DAMPs which may transfer to nearby dendritic cells and induce their maturation into CXCL16-positive antigen-presenting cells (APCs). Next, these APCs migrate into peripheral lymph nodes and spleen to recruit CXCR6 + NKT cells to enhance inflammation. Abbreviations: APM, arrector pili muscle; CUMS, chronic unpredictable mild stress; DC, dendritic cells; DAMPs, damage-associated molecular patterns; HFSC, hair follicle stem cell; KC, keratinocytes; McSC, melanocyte stem cell; M1, macrophages M1 pro-inflammatory phenotype; NKT, natural killer T cells; SG, Sebaceous gland; Tem, effector memory T cells
Comments (0)