2.1 Study Design and Participants
This randomized, parallel-group, open-label, phase I study (NCT05146869) was conducted in China. Adults (aged between 18 and 75 years) diagnosed with type 2 diabetes according to the World Health Organization 1999 criteria were enrolled. Patients had HbA1c between 7.0 and 9.5% and a body mass index within the range of 19–35 kg/m2. Key exclusion criteria included fasting plasma glucose > 13.9 mmol/L, requirement of insulin treatment based on the investigator’s judgement, acute or serious complications of diabetes, a history of serious hypoglycemia, and inadequate organ function. The full inclusion and exclusion criteria are provided in the Electronic Supplementary Material (ESM) pages 2–4.
The study consisted of screening, run-in, baseline, treatment (days 1–9), and follow-up (days 9–11) periods. Eligible patients were required to have a run-in period of at least 1 week when patients followed the diet and exercise routines recommended by the investigators. After completing the run-in phase, patients’ eligibility was confirmed in the baseline period, and they then remained in the research units throughout the treatment and follow-up periods (days 1–11).
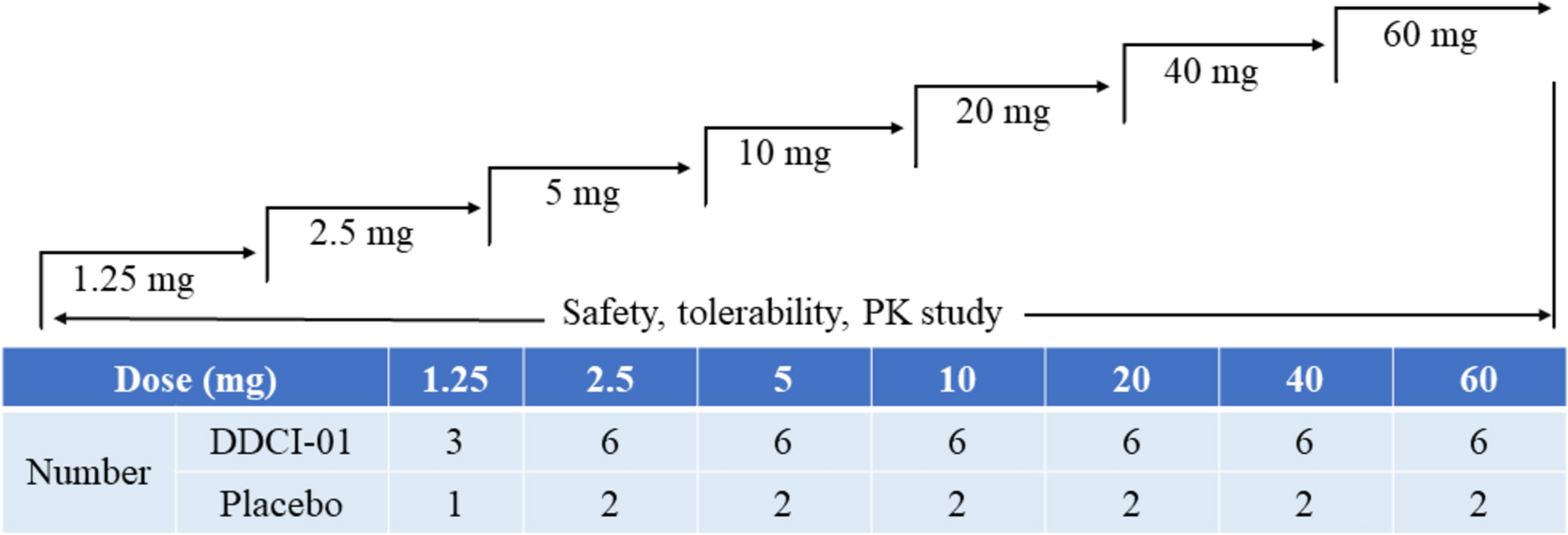
Enrolled patients were assigned 1:1:1 to the DBPR108 50-mg, 100-mg, or 200-mg groups using the block randomization method. DBPR108 tablets were administered orally at a fasting state, once daily at the same time, on days 1 and 3–9, for a total of eight doses. Block randomization was employed with a fixed block size of six to ensure balanced group assignments throughout the study. The randomization sequence was generated by an independent statistician using SAS version 9.4, and allocation was implemented through an interactive response technology system.
The study followed the principles of the Declaration of Helsinki and requirements of Good Clinical Practices and applicable regulations. The study protocol was approved by the Ethics Committee for Clinical Trials of Beijing Anzhen Hospital, Capital Medical University, and the Ethics Committee of The First Affiliated Hospital of Bengbu Medical University. All patients provided written informed consents for their study participation.
2.2 PK Assessment
Blood samples to determine DBPR108 concentration were collected from each participant at the prespecified time points: pre-dose; 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, 24, 36, and 48 h post-dose on days 1 and 9; and pre-dose on days 7 and 8. Blood samples were collected in heparin sodium-containing tubes and were centrifuged at 1700 ± 50 g at 4 °C for 10 min. Then 50% acetic acid solution was added to the isolated plasma in a 1:100 volume ratio before storing at ≤ – 60 °C until analysis.
Plasma concentration of DBPR108 was determined using a validated liquid chromatography with tandem mass spectrometry method at Nanjing Clinical Tech Laboratories Inc. (Nanjing, China). The analytic range was 1.00–2000 ng/mL with a lower limit of quantitation (LLOQ) of 1.00 ng/mL.
2.3 PD Assessment
Blood samples for determination of DPP-4 activity and active GLP-1 level were collected at pre-dose; 1, 2, 4, 6, 8, 12, 24, 36, and 48 h post-dose on days 1 and 9; and pre-dose on days 7 and 8.
Blood samples were collected in K2EDTA-containing tubes and P800 blood collection tubes for the detection of DPP-4 activity and active GLP-1 level, respectively. Blood samples were centrifuged at 1300 ± 50 g at 2–8 °C for 10 min, and the resulting plasma samples were stored at ≤ – 60 °C until analysis.
Activity of DPP-4 was determined using the EnVision HTS Microplate Reader (PerkinElmer, MA, USA) with a LLOQ of DPP-4 activity at 21.23 μU/mL. Active GLP-1 level was determined by the MSD® V-PLEX GLP-1 Active Kit (Meso Scale Discovery, MD, USA) with a LLOQ of 0.182 pM. Bioanalyses of DPP-4 and GLP-1 were carried out by Accurant Biotech (Ningbo, China).
2.4 Safety Assessment
Safety evaluation included measurements of vital signs, physical examinations, electrocardiogram, and laboratory tests. Patients were monitored for adverse events from screening to study completion or early withdrawal.
Adverse events were coded in accordance with Medical Dictionary for Regulatory Activities Terminology version 23.0, and severity was graded as mild, moderate, or severe.
2.5 Statistical Analysis
The primary endpoints included PK and PD characteristics after a single dose and multiple doses of DBPR108. Safety was evaluated as a secondary outcome. According to the National Medical Products Administration guidelines [19, 20], and on the basis of the experience of a previous study [21], phase I studies generally include 8–12 patients in each group. This study planned to enroll ten patients in each group.
Demographic and baseline characteristics were summarized in the full analysis set (FAS), which included all randomized patients who received at least one dose of DBPR108. Safety was evaluated in the safety set (SS), which included patients who received at least one dose of DBPR108, and patients in the SS were grouped according to the actual dose level they received. The PK concentration set (PKCS) included all randomized patients who had received DBPR108 and had at least one measurable PK concentration. PK parameters were calculated from the PK parameter set (PKPS), which included patients who were randomized, had received DBPR108, and had at least one evaluable PK parameter. Patients who deviated from the protocol or used prohibited concomitant medications that would influence PK parameters were excluded from the PKPS. PD was analyzed in the PD set (PDS), which included all patients in the SS who had at least one evaluable PD assessment.
PK concentrations and plasma concentration–time profiles were summarized by treatment group in the PKCS. PK parameters including maximum plasma drug concentration (Cmax), area under the concentration–time curve from time zero to infinity (AUCinf), AUC from time zero to the time of the last measurable concentration (AUClast), AUC during a dosage interval (AUCtau), terminal elimination rate constant (λz,), time to reach the maximum plasma concentration (Tmax), apparent volume of distribution during the terminal phase (Vz/F), elimination half-life (t½), apparent total body clearance for extravascular administration (CL/F), maximum plasma drug concentration at steady state (Cmax,ss), minimum steady-state plasma drug concentration during a dosage interval (Cmin,ss), AUC from time zero to infinity at steady state (AUCinf,ss), AUC from time zero to the time of the last measurable concentration at steady state (AUClast,ss), AUC during a dosage interval at steady state (AUCtau,ss), time to reach the maximum plasma concentration at steady state (Tmax,ss), elimination half-life at steady state (t½,ss), apparent total body clearance for extravascular at steady state (CLss/F), and accumulation ration (Rac) were calculated from individual plasma concentration and actual sampling times using noncompartmental analysis in the PKPS.
PD analyses of DPP-4 inhibition and active GLP‐1 level were summarized descriptively in the PDS. DPP-4 inhibition was defined as the percentage of change in DPP-4 activity from baseline. Relationships between PK parameters and indicators of PD were analyzed using linear regression and Emax models.
PK parameters were calculated using Phoenix WinNonlin version 8.3 (Certara USA, Inc.). All other analyses were conducted using SAS version 9.4.


Comments (0)