2.1 Patients and Study Design
This study was a prospective, open-labelled, multi-centre, observational study performed at four Australian hospitals between July 2021 and August 2022. Patients over 18 years old admitted to hospital with confirmed SARS-CoV-2 infection (nucleic acid amplification test positive within 14 days of symptom onset) were eligible for inclusion if they were prescribed remdesivir for treatment of COVID-19. Informed consent was obtained from all participants, or their responsible person, prior to enrolment. Data on patient demographics (age, sex, weight, height, body mass index (BMI) and ethnicity), clinical characteristics (comorbidities, smoking status, oxygenation, ventilation status, medications, antibiotic use, antiviral use, immunomodulatory agents and sepsis organ function assessment (SOFA) score) and clinical laboratory investigations (aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), gamma-glutamyl transferase (GGT), platelet count, white cell count, albumin, and bilirubin and serum creatinine concentrations) were collected from the patient’s medical record. Estimated glomerular filtration rate (eGFR) was calculated on the basis of serum creatinine concentrations using the chronic kidney disease epidemiology collaboration formula (CKD-EPI) formula [13], with appropriate caution being applied in patients with significant acute kidney injury [14]. Patients with eGFR < 30 mL/min/1.73 m2 were excluded from this pharmacokinetic study.
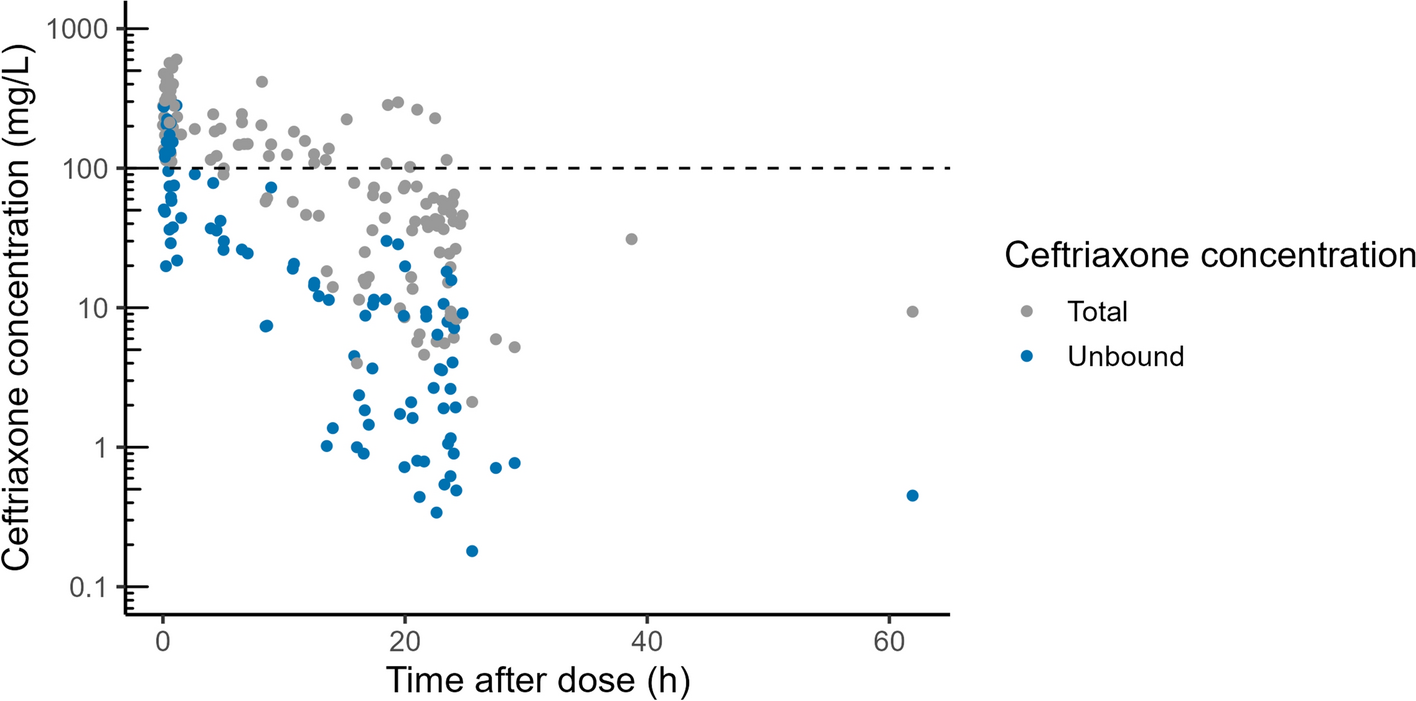
Remdesivir was administered as an intravenous infusion over 60 min at a loading dose of 200 mg on day 1 followed from day 2 up to day 5 or day 10 as a 100 mg dose once daily over 60 min. Blood samples were collected in sodium fluoride/potassium oxalate anticoagulant tubes (for measuring remdesivir and GS-441524 concentrations) or potassium EDTA (if measuring GS-441524 concentrations only). Where possible, multiple samples were collected over one dosing interval on two separate occasions. Occasion 1 occurred on day 1–3 of remdesivir therapy and occasion 2 occurred on a different day between days 3–7 of therapy. Samples were collected at pre-dose, 1.5, 3, 6, 8–12 and 20–24 h after commencement of infusion. Surplus plasma from samples obtained for clinical care at other times during the dosing interval were also obtained, where possible.
Samples for measuring remdesivir and GS-441524 were placed in an ice water bath immediately on collection owing to the instability of remdesivir at room temperature [15, 16]. Samples were centrifuged within 1 h of collection and 1–2 mL plasma were transferred to polypropylene cryovial for storage at –80 °C for later drug analysis. Surplus plasma samples for measuring GS-441524 (GS-441524 is stable at room temperature for 48 h [17]) were stored refrigerated in the pathology laboratory until being processed within 48 h of collection.
2.2 UHPLC-MS/MS Assay
Plasma concentrations of remdesivir and GS-441524 were determined by an ultra-high performance liquid chromatography coupled with tandem mass spectrometry (UHPLC-MS/MS) method. Chromatographic separation was achieved with a Kinetex C8, 100 mm × 2.1 mm (1.7 μm) analytical column (Phenomenex, Torrence, USA) preceded by C8 UHPLC guard cartridge (Phenomenex, Torrence, USA). Mobile phase A was 0.1% formic acid in water (v/v), and mobile phase B was 0.1% formic acid in acetonitrile (v/v). Mobile phase was delivered as a gradient from 30 to 90% of mobile phase B over 6 min at a flow 0.30 mL/min. Remdesivir and GS-441524 were monitored by positive-mode electrospray at multiple reaction monitoring (MRM) of m/z 603.20→229.05 and 291.95→163.15, respectively. The lower limit of quantification (LLOQ) was 5 ng/mL for both remdesivir and GS-441524. Test samples were assayed in batches alongside calibrators and quality controls and the results were subject to pre-established batch acceptance criteria [18].
2.3 Population Pharmacokinetic Analysis
Patients contributing plasma concentrations were divided into model-building and external validation datasets at an approximate ratio of 3:1, prioritising patients with complete data collection to the model-building cohort. The non-linear mixed-effects modelling program Monolix version 2021R2 (Lixoft SAS, a Simulations Plus company, Antony, France), implementing the stochastic approximation expectation maximization (SAEM) algorithm, was used. An integrated model containing both remdesivir and its metabolite GS-441524 was developed. To account for the differences in molecular weight (MW) between remdesivir (MW = 602.585) and GS-441524 (MW = 291.26), concentration data were converted from ng/mL to µM.
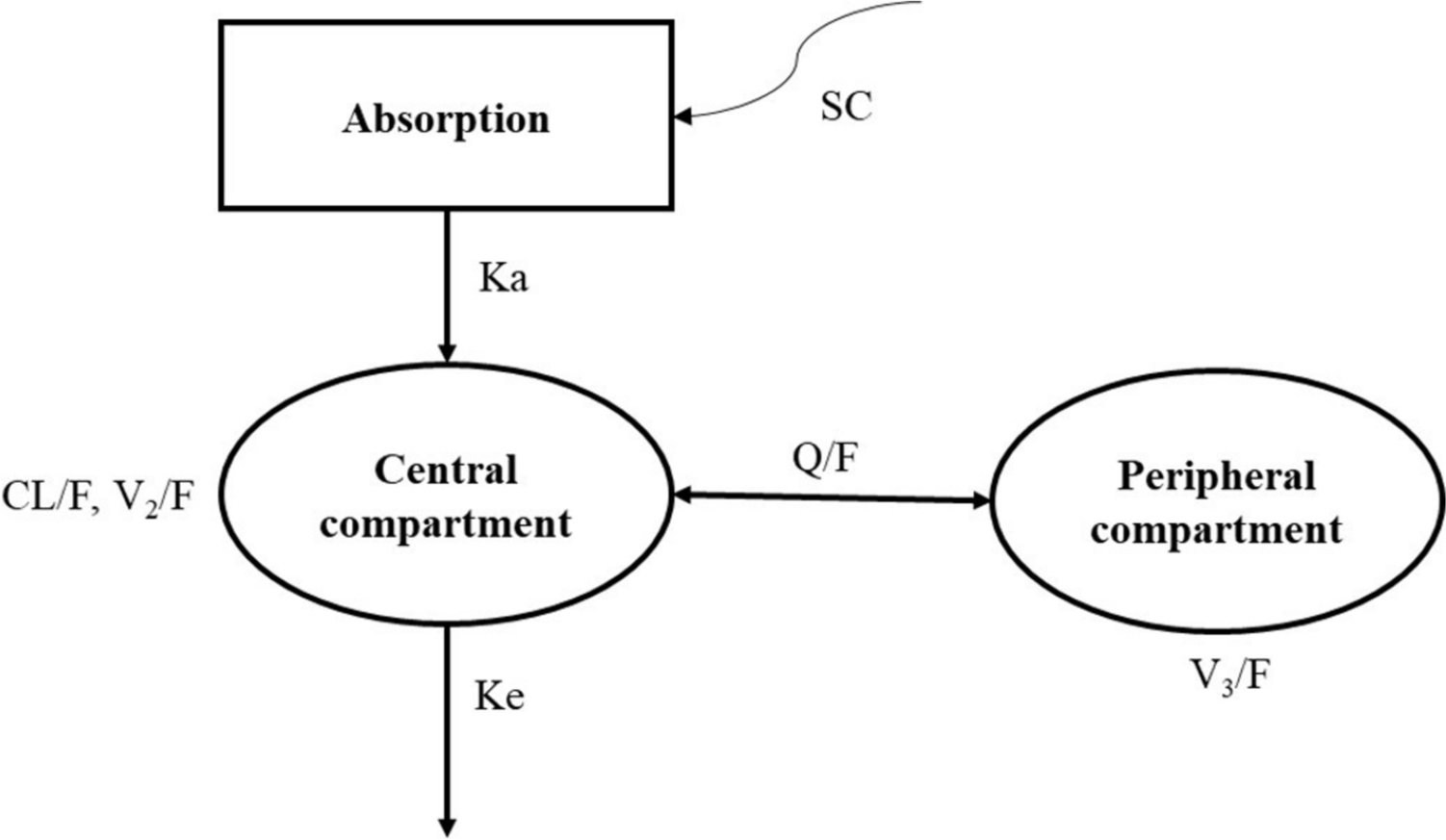
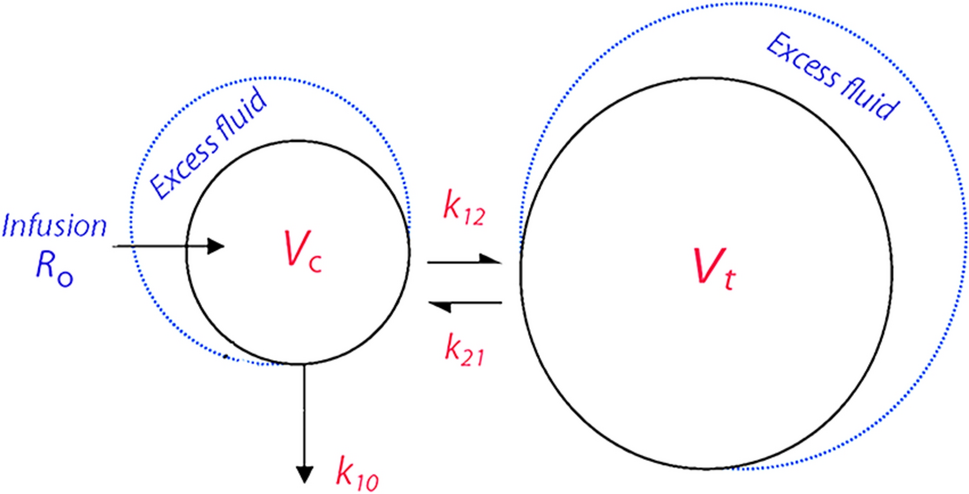
One-, two- and three-compartment models with first order elimination were tested as base structural models for remdesivir and GS-441524. As 10% of the administered remdesivir is excreted unchanged in the urine [19], renal clearance of remdesivir was fixed to 10% of the total remdesivir clearance. All individual parameters were assumed to be log-normally distributed. The between-subject variability (BSV or ω) was described using an exponential model. To describe the residual variability (ε), several error models (constant, proportional or combined error model) were tested. The most appropriate model was selected on the basis of the following criteria: the objective function value (OFV) (defined as –2 × log-likelihood, –2LL), usual goodness-of-fit (GOF) plots, and relative standard error (RSE) of parameter estimates.
2.4 Covariate Analysis
From the base model, the effects of the following covariates on remdesivir and GS-441524 pharmacokinetic parameters, with consideration of biological plausibility, were evaluated: age, gender, height, body weight, BMI, eGFR, SOFA score, serum albumin, bilirubin, ALT, AST, ALP and GGT level. Continuous covariates were modelled using linear and power functions.
The covariate model was built using a stepwise procedure with forward inclusion and backward elimination. The statistical significance of each covariate was individually evaluated during the stepwise deletion using the likelihood ratio test (LRT). The covariate was retained in the model if the effect was biologically plausible, it produced a reduction in BSV of the parameter and the OFV was decreased by at least 3.84 (P < 0.05) when included (as compared with the base model) and increased by 6.63 (P < 0.01) when eliminated (as compared with the model with the covariate included).
2.5 Model Evaluation
Evaluation of the model was based on goodness-of-fit (GOF) plots, including observations versus individual and population predictions and plots of normalized prediction distribution error (NPDE) versus population predictions and time. A visual predictive check (VPC) was performed using 500 simulations with the final model. This plot shows the time course of the 5th, 50th and 95th percentiles of the simulated profiles, compared with observed data.
The accuracy of the final model was also examined using a bootstrap method. A 1000-run bootstrap resampling procedure was performed in Monolix using the Rsmlx (R Speaks ‘Monolix’, version 4.0.2) package in R software (version 4.1.3). The median, 2.5% and 97.5% values obtained from the 1000 bootstrap runs for each parameter were calculated and compared with the estimates from the original data.
2.6 External Validation
GS-441524 concentrations from a sub-set of patients were used for external validation to assess the predictive performance of the final model. The relative prediction error (PE, Eq. 1) of the model was estimated by comparing the population predicted concentrations (Cpred) and the corresponding observations (Cobs) for each subject at each time point in the external validation dataset. Mean prediction error (MPE) and the root mean square error (RMSE, Eq. 2) were calculated to assess the prediction bias and precision. Bland–Altman plots were developed to visualize the trends of bias. The population pharmacokinetic model was regarded as valid when both the mean and median values of PE were less than 20% [20].
$$PE = \left(\frac_} -_}}_}}\right)\times 100,$$
(1)
$$RMSE=\sqrt\sum ^}.$$
(2)
2.7 Dosing Simulations
Monte Carlo simulations (n = 1000) were performed by Simulx version 2021R2 (Lixoft SAS, a Simulations Plus company, Antony, France) using the final pharmacokinetic model to generate pharmacokinetic profiles of remdesivir and GS-441524 for a 5-day remdesivir treatment regimen. A typical virtual patient with COVID-19 with age of 70 years and an eGFR of 80 mL/min/1.73 m2 was used to simulate the pharmacokinetic profiles of remdesivir and GS-441524 for six 5-day remdesivir dosing regimens: (1) currently licensed dosage of 200 mg loading dose followed by 100 mg once daily; (2) a loading dose of 200 mg followed by 150 mg once daily; (3) a 200 mg loading dose on day 1 followed by 100 mg every 12 h; (4) a loading dose of 100 mg followed by 50 mg every 6 h for 5 days in total; (5) a loading dose of 300 mg followed by 50 mg every 6 h for 5 days in total as recommended by others[21]; and (6) 200 mg dose on day 1 and day 2. All doses were simulated as a 1 h intravenous infusion and were chosen to explore highly varied dosing regimens which may inform alternative and potentially more effective dosing regimens. To investigate the effect of the eGFR and age on GS-441524 pharmacokinetics, simulations were performed on the basis of different eGFRs (40, 80, 120 mL/min/1.73 m2) and ages (30, 70 and 90 years). The standard licensed dosing regimen was used for these simulations.
As the clinical pharmacodynamics of remdesivir have not been described, we used two target exposures, an unbound plasma concentration above the in vitro half-maximal effective concentration (EC50) and above the 90%-effective concentration (EC90) for remdesivir and GS-441524. Literature-reported EC50 or EC90 values for remdesivir and GS-441524 are summarised in the Supplementary Material Table S2. These values were corrected for protein binding, 88% for remdesivir and 2% for GS-441524, to compare directly with measured plasma concentrations of remdesivir or GS-441524 [22].
2.8 Ethics Approval
The study protocol was approved by Human Research Ethics Committees of St Vincent’s Hospital Sydney (2021/ETH11567), The University of Queensland (2022/HE00260) and Royal Brisbane and Women’s Hospital (HREC/2020/QRBW/66533).


Comments (0)