
Remember me





Two human iPSC lines, CD05 and CD07 (full IDs: CD0000005 and CD0000007), were used for CRISPR/Cas editing. These two iPSC lines were initially derived from cryopreserved lymphocytes (CPLs) using the genome-integration-free Sendai virus method (Cytotune Sendai Virus 2.0; Invitrogen) at the Rutgers University Cell and DNA Repository (RUCDR) (also known as NIMH Stem Cell Center and Infinity Biologix, currently Sampled). The two iPSC lines have been used in our previous ASoC mapping studies [35,36,37]. The two donors are all healthy control males of European ancestry aged 65 and 59 for lines CD05 and CD07, respectively. The donors were also analysed for copy number variants (CNVs), and none have large CNVs (> 100 kb) [38]. They were heterozygous (T/C) at SNP site rs1532278, and the two iPSC lines were CRISPR/Cas9-edited to homozygous T/T and C/C. Two to three clones per genotype were obtained. Quality control measures for the unedited and edited iPSC lines included IF staining for pluripotency, mycoplasma contamination test, RNA-seq-based pluripotency test (Pluritest), and eSNP-karyotyping as previously described [35, 36]. The Endeavor Health (formerly NorthShore University HealthSystem) institutional review board (IRB) approved this study.
For iPSC maintenance, cells were cultured using mTeSR Plus media (StemCell Technologies, #100–0276), with medium changes performed every other day, and passaged as clumps every 4–6 days using ReLeSR (StemCell Technologies, #100–0483).
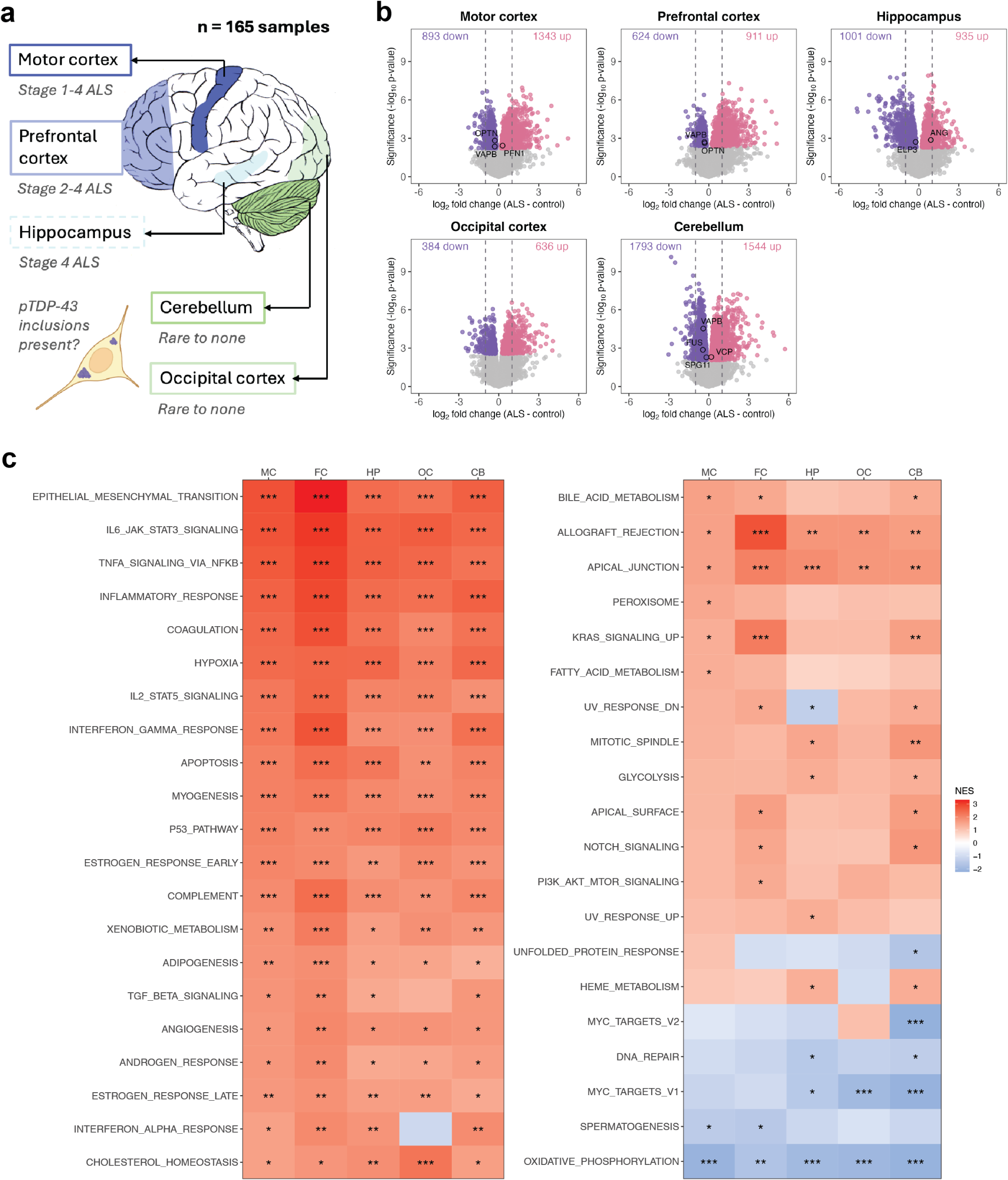
iPSC differentiation into different brain cell types for ASoC mappingTo generate ATAC-seq data and map ASoC SNPs in different brain cell types, we have previously generated human iPSC-derived microglia (iMG) (n = 38 donors) [37], astrocytes (iAst) (n = 18 donors) [37], glutamatergic (iGlut, n = 36) [35,36,37], GABAergic (iGABA, n = 30) [35,36,37], and dopaminergic neurons (iDN, n = 39) [35,36,37]. The detailed methods for generating these cell types have been previously described [37]. The purity of iMG (TREM2+CD45+PU1+) and iAst (Vimentin+/GFAP+/S100β+) is > 90% [37]. The purity of iGlut (VGLUT1+/MAP2+), iGABA (GAD1+ or GAD2+/MAP2+) and iDN (HT+/MAP2+) is > 85% [35, 36]. The transcriptomic (using RNA-seq data) and/or epigenomics (using ATAC-seq) similarity of each of these iPSC-derived cell types to corresponding brain cell types have been previously confirmed by principal components analyses [35,36,37].
Bulk ATAC-seq and ASoC SNP callingATAC-seq sample preparation was performed as previously described [35, 36]. Briefly, 75,000 viable cells were used for each transposition mixture reaction. Samples were then incubated at 37 °C for 30 min on a thermomixer at 1,000 rpm. The eluted DNA was shipped to the University of Minnesota Genomic Center for library preparation and ATAC-seq. All raw sequence reads generated by Illumina NextSeq were demultiplexed at the University of Minnesota Genomics Center and provided as 2 × 75 bp paired-end FASTQ files (targeting 60 M reads per sample). Only paired-end reads that survived Trimmomatic processing v0.39 (ILLUMINACLIP:NexteraPE-PE.fa:2:30:7, SLIDINGWINDOW:3:18, MINLENGTH:26) were retained. The sequencing reads mapping and the preparation of cell line-specific VCF files for OCR peak and ASoC SNP calling were previously described in detail [37]. All analyzed ATAC-seq samples passed standard QC based on the characteristic nucleosomal periodicity of the insert fragment size distribution and high signal-to-noise ratio around transcription start sites (TSS) [37].
For ASoC mapping, GATK (version 4.1.8.1) was used as recommended by the GATK Best Practices (software.broadinstitute.org/Gatk/best-practices/) [39] as described previously in detail [37]. Briefly, from individual VCF genotype files, heterozygous SNP sites with tranche level > 99.5% were extracted. Biallelic SNP sites (GT: 0/1) with minimum read depth count (DP) ≥ 20 and minimum reference or alternative allele count ≥ 2 were retained. We then compared the combined allelic counts of each allele of each SNP across all heterozygous samples. The binomial p-values (non-hyperbolic) were calculated using the binom.test(x, n, P = 0.5, alternative = “two.sided”, conf.level = 0.95) from the R package. Benjamini & Hochberg multiple-testing correction was applied to all qualified SNPs. We called ASoC SNP using FDR value = 0.05 as a cut-off. We have previously verified that the ASoC SNP calling is not affected by donor’s sex, age or caseness [35, 36]. Based on power calculations [35, 36], we expected to have sufficient power to call ASoC SNPs even for iAst data from the smallest number of donor lines (n = 18).
CRISPR-cas9 editing and Sanger sequencing confirmationThe online tool Benchling (www.benchling.com) was used to design CRISPR guide RNA (gRNA), and the gRNAs with highest off-target score (specificity) were selected (Table S15). The gRNAs were cloned into CROPseq-Guide-Puro vector (Addgene, #86,708) [40]. The gRNA plasmid DNAs, together with the plasmid DNAs of pSpCas9(BB)− 2A-Puro (Addgene, #62,988) [41] and pSUPERIOR.puro-shp53 (Addgene, #38,035) (included to transiently inhibit p53 and increase editing efficiency [42]) were transiently transfected into iPSCs. For transfection, iPSCs were dissociated into single cells using accutase (StemCell, 07920) in a 15 ml centrifuge tube at a density of 4–6 × 105/1.8 ml in the presence of 10 μM ROCK inhibitor (ROCKi, R&D Systems, 1254/1). Lipofectamine stem reagent (Thermo Fisher Scientific, STEM00001) was used for transfections. DNA including pSpCas9(BB)− 2A-Puro (0.5 μl from 1 μl/ug stock), CROPseq-Guide-Puro vector (1 μl from 1 μl/ug stock), pSUPERIOR.puro-shp53 vector (1 μl from 1 μl/ug stock) and ssODNs carrying the desired SNP allele (6 μl from 100uM stock) were mixed into 100 μl Opti-MEM media (Thermo Fisher Scientific, 31,985,062) in a 1.5 ml tube. Another 1.5 ml tube was filled with 8 μl Lipofectamine stem reagent and 100 μl Opti-MEM media. The reagents in the two 1.5 ml tubes were combined and added into iPSC single cell suspension, followed by gently mixing and replating into one well of a 6-well plate. Sixteen hours post-transfection, the medium was replaced with fresh medium containing 0.25 μg/ml puromycin (Thermo Fisher Scientific, A1113802) and 10 μM ROCKi. Forty-eight hours post transfection, the medium was refreshed with the same reagents. Seventy-two hours post transfection, the puromycin-containing medium was removed, and the cells were cultured in fresh medium with 5 μM ROCKi. Half-medium (without ROCKi) change was made every 2–3 days until iPSC colonies were formed. 10–14 days post transfection, iPSC colonies were individually picked into 96-well plates. DNA from a small fraction of cells in each colony was then extracted (Epicentre, QE09050) and used for Sanger sequencing confirmation of accurate editing. Subcloning with at a low density (2–5000 cells on 6-cm dish) was carried out to ensure an iPSC clone was pure. Following successful editing, three predicted top-ranking off-target editing sites were subjected to Sanger sequencing to confirm the absence of off-target editing (Table S15 and Fig. S2 A).
For OCR deletion, up- or downstream gRNAs were cloned into the vector pSpCas9(BB)− 2A-Puro to co-express gRNAs and Cas9. Transfection was conducted as described above. The presence of on-target OCR deletion and the absence of the predicted off-target editing in the selected iPSC clones were confirmed by Sanger sequencing.
Preparation of primary mouse astrocytesPrimary mouse astrocytes were extracted as previously described [43]. Briefly, the brains were harvested from Day 0–2 pups of C57BL/6 J. Their meninges were removed, minced and centrifugated in cold Hanks'balanced salt solution (HBSS buffer, Thermo Fisher Scientific, 88,284). Then, tissue pellets were resuspended, dissociated, and filtered through a cell strainer to generate a single cell suspension. All cells were seeded into T- 75 flasks, maintained in DMEM (Thermo Fisher Scientific, 10,569,016) with 10% FBS (Thermo Fisher Scientific, A5209501). The obtained astrocytes were used within a month (≤ 4 cell passages). All astrocytes used in this study were from the same extraction. For each assay, the astrocytes were recovered from the same liquid nitrogen stock and used with the same cell passages to ensure the consistency.
Glutaminergic neuronal (iGlut) differentiationiGlut neurons were differentiated from iPSC according to the previous protocols [36, 44] with some modifications. Briefly, on day − 1, iPSCs were dissociated with accutase and placed in 6-well plates at a density of 5 × 105/well in the presence of 5 μM ROCKi. On day 0, NGN2 and rtTA lentivirus were added into mTeSR Plus medium with 5 μM ROCKi to infect iPSCs. On day 1, Neurobasal media (Thermo Fisher Scientific, 21,103,049) containing 1 × B27 supplement (Thermo Fisher Scientific, 17,504,044), 1 × GlutaMax (Thermo Fisher Scientific, 35,050,061), 5 μg/ml Doxycycline (Dox, Sigma, D9891) and 5 μM ROCKi were used to initiate the differentiation. 1 μg/ml puromycin selection was performed from Day 2 to Day 4. On day 5, the cells were dissociated with accutase, replated into matrigel-coated plates, and maintained in Neurobasal media supplemented with 1 × Glutamax, 1 × B27, 5 µg/ml doxycycline, 10 ng/ml BDNF/GDNF/NT- 3 (Pepro Tech, 450–02/450–10/450–3), and 5% FBS. On day 6, the culture medium was refreshed with the same one used on Day 5 without FBS, and 1 µM Ara-C (Millipore-Sigma, C6645) was applied to the medium and kept for two days to ensure the purity of postmitotic neurons. On day 8, Ara-C was withdrawn and the neurons were maintained with one half-medium change every 3 days until they were mature enough for the assays.
For CLU and LDs staining in pure iGlut culture, day- 5 neurons were directly replated onto 12 mm glass coverslips (Neuvitro, GG- 12–15-Pre) at 50,000 cells/coverslip. Neurons were maintained on the coverslip until day 25 by refreshing half the medium every three days. For neuron morphology and LD staining in the iGlut-mAst co-culture system, 100,000 astrocytes were first placed onto glass coverslips 24 h before dissociating neurons in 24-well plates with DMEM medium with 10% FBS. Then, day- 8 iGlut neurons were dissociated with accutase, suspended in Neurobasal medium, and seeded on top of the layer of astrocytes at 300,000 cells/well after removing the DMEM medium. On day 9, one-half of the medium was replaced without FBS, and on day 10, all medium were refreshed to remove FBS completely. Thereafter, all medium was refreshed every 3 days until day 30. For ChIP-qPCR and RNA seq purposes, day- 8 iGlut and mAst were dissociated and placed together at appropriate dishes/plates, and the cultures were maintained the same way as iGlut-mAst co-culture described above.
iPSC differentiation into astrocytes (iAst)iAst were differentiated from iPSC based on the NgN2 method [45] with minor modifications. Briefly, on day − 1 and day 0, the same iGlut differentiation procedures were followed. On day 2, a cocktail of SB431542 (10 µM) (Tocris Bioscience, 1614), XAV939 (2 µM) (Tocris Bioscience, 3748), LDN193189 (100 nM) (Tocris Bioscience, 6053), Dox (5 µg/ml) and ROCKi (5 µM) were added into N2 medium that was constituted with DMEM/F12 (Gibco, 11,320–033), 1 × Glutamax, 0.3% Sucrose (Millipore-Sigma, S0389), and 1 × N2 supplement B (StemCell Technologies, 07156). On day 3, SB431542 (5 µM), XAV939 (1 µM), LDN193189 (50 nM), Dox (5 µg/ml) and Puromycin (5 µg/ml) were added into N2 medium to start cell selection. On day 4, the culture medium was refreshed with N2 medium supplemented with Dox (5 µg/ml) and Puromycin (5 µg/ml). On day 5, all cells were dissociated with Accutase, suspended with Astrocyte medium (SiceneCell, 1801) containing 5 µM ROCKi, and replated onto Matrigel-coated 10-cm dishes. Cells were passaged at 1:2 ratio in Astrocyte medium every 3–4 days. On day 25, the cells were fixed with 4% PFA, permeabilizated with 0.1% Triton X- 100 (Sigma, SLCD3244) and stained with S100β (Millipore-Sigma, S2532, Mouse, 1:500) and Vimentin (Cell Signaling, D21H3, Rabbit, 1:200) to determine iAst purity (~ 100%).
iGlut morphology and CLU stainingDay- 30 iGlut-mAst co-culture on coverslips were fixed with 4% PFA for 15 min and permeabilized with 0.1% Triton X- 100 for 20 min. The fixed cells were incubated with antibodies again PSD- 95 (NeuroMab, clone K28/43, Mouse, 1:1,000), SYP (Abcam, ab32127, Rabbit, 1:500), and MAP2 (Synaptic System, 188,004, Guinea pig, 1: 1000) overnight at 4 °C. After three washes with PBS, the corresponding secondary antibodies including Donkey anti-Mouse Alexa 488 (Thermo Fisher Scientific, A21202, 1:200), Donkey anti-Rabbit Alexa 594 (Thermo Fisher Scientific, A21207, 1:200), and Goat anti-Guinea Pig Alexa 647 (Thermo Fisher Scientific, A21450, 1:200) were added and further incubated for two hours at room temperature. Nuclei were stained with DAPI (0.5 mg/ml) for 2 min at room temperature before the coverslips were mounted (Fluorescent Mounting Medium, Millipore-Sigma, F4680). For IF staining of neuron identity, the cultures were stained with HuNu (Millipore-Sigma, MAB1281, Mouse, 1:200), GFAP (DAKO, Z0334, Rabbit, 1:500), and MAP2 (Synaptic System, 188,004, Guinea pig, 1: 1000) antibodies. For iGlut purity quantification assay (Fig. S1D)., the cultures were stained with HuNu (Thermo Fisher Scientific, RBM5 - 346-P1, Rabbit, 1:100), MAP2 (Millipore-Sigma, AB5543, Chicken, 1:5000), vGlut1 (Synaptic System, 135,511, mouse, 1: 100), and Ki67 (Thermo Fisher Scientific, 740008 T, Rat, 1: 500) antibodies. All antibodies were dissolved in PBS with 1% BSA (Thermo Fisher Scientific, 15,260,037) and 0.1% Triton X- 100.
For CLU staining in pure culture iGlut, cells were fixed on day 25, permeabilized, and incubated with CLU (Abcam, ab69644, Rabbit, 1:200) and NeuN (Millipore-Sigma, MAB377, Mouse, 1:50) antibodies. NeuN immunostaining was used to visualize perikaryon where most CLU signals were located (in the Image quantification section below). All procedures were the same as above.
RNA isolation from cell cultures for qPCR and RNA-seqTotal RNA was extracted using the RNeasy Plus Kit (Qiagen, 74,134). Briefly, all cells were directly lysed in RLT plus buffer, further separated and eluted in RNase-free water. For RNA-seq, all RNA samples were sequenced on the Illumina NovaSeq 2000 platform with paired-end reads (2 × 150 bp) (Novogene). For qPCR, RNAs were first reversed transcribed to cDNAs using a high-capacity cDNA reverse transcription kit (Applied Biosystems, 4,368,814) and further amplified with TaqMan Universal PCR Master Mix (Applied Biosystems, 4,364,338) on a Roche 480 II instrument. All qPCR primers were listed in Table S15.
Chromatin immunoprecipitation (ChIP) qPCRChIP-qPCR assay was performed following the protocol of Magna ChIP A/G Chromatin Immunoprecipitation Kit (Millipore-Sigma, 17–10,085). Briefly, 1% formaldehyde (Thermo Fisher Scientific, 28,908) was directly added into the medium of day- 30 iGlut-mAst co-cultures to cross-linking proteins to DNA. Then, glycine was added to terminate the cross-linking. Cells were harvested in ice-cold PBS, pelleted, sonicated, and immunoprecipitated with 10 µg ISL2 (R and D Systems, AF4244, Sheep) or DRGX antibodies (Bioss, bs- 11827R, Rabbit) and Protein A/G magnetic beads overnight at 4 °C. Protein A/G magnetic beads with TF-binding DNAs were separated on a magnetic stand. TF-DNA complexes were eluted in an elution buffer with proteinase K at 62 °C with 2 h of shaking, followed by a 95 °C incubation for 10 min to denature proteinase K. Lastly, the released DNAs were purified for qPCR. About 2% of sample input after sonication without immunoprecipitation was used to normalize the loading input. Normal sheep IgG (R&D Systems, 5–001-A) or rabbit IgG (Cell Signaling Technology, 2729) were used as negative control to exclude non-specific binding of antibodies. The ChIP-qPCR primers are listed in Table S15.
ISL2 siRNA knockdowniGlut were directly replated onto a 24-well plate at 500,000 cells/well on Day 5. On day 30, the neurons were transfected either by 50 nM human ISL2 siRNA (Horizon Discovery Biosciences, L- 016725–00–0005) or 50 nM non-targeting control siRNA (Horizon Discovery Biosciences, D- 001810–10 - 05) following the protocol of Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific, 13,778,030). 72 h post transfection, RNAs were extracted to quantify ISL2 and CLU mRNA levels by qPCR. The qPCR primers are listed in Table S15.
Enzyme-linked immunosorbent assay (ELISA) assayFor both iGlut pure culture and iGlut-mAst co-culture, the culture medium was completely refreshed on day 26 and further collected on day 30. The conditioned media were centrifuged for 10 min with 3,000 rpm at 4 °C to remove the cell debris. Cells were harvested and protein quantification was performed by the BCA method to normalize the ELISA detection from cell lysis or the supernatant. ELISA quantifications of human CLU (R&D system, DCLU00, specific for human CLU), human Aβ 1–40 (R&D system, DAB140B), human Aβ 1–42 (R&D system, DAB142), and mouse CLU (R&D system, MCLU00, specific for mouse CLU) were performed according to the vendors’ protocol.
Western blottingWestern blotting was performed as previously described [43]. Briefly, cells were lysed in NP- 40 lysis buffer (Thermo Fisher Scientific, J60766-AK) with proteinase inhibitors (Roche, 04693159001) and phosphatase inhibitors (Roche, 04906845001). The cell lysates were sonicated and denatured in Laemmli Sample Buffer (Bio-Rad, 1,610,747). BCA kit (Thermo Fisher Scientific, 23,225) was used for total protein quantification. The cellular protein extracts were fractioned through 10% homemade SDS-PAGE gels, transferred to a PVDF membranes (Bio-Rad, 1,620,177) and subsequently immunoblotted. The primary antibodies included PSD- 95 (Synaptic System, 124,011, mouse, 1:1,000), SYP (Abcam, ab32127, Rabbit, 1:2,000), and β-actin (Cell Signaling Technology, 3700, 1:5,000). The secondary antibodies included anti-rabbit-HRP (Cell Signaling Technology, 7074, 1:5,000) and anti-mouse-HRP (Cell Signaling Technology, 7076, 1:5,000). All Western blotting images were quantified using FIJI [46], and specific protein signals were normalized to corresponding β-actin signals in each sample.
Calcium imagingiGlut were infected with AAV-GCaMP6 m (pAAV.Syn. GCaMP6 m.WPRE.SV40, Addgene, 100,841-AAV9) or AAV-jRCaMP1b (pAAV.Syn.NES-jRCaMP1b.WPRE.SV40, Addgene, 100,851-AAV9) at 105 MOI on day 8. The infected neurons were replated together with mAst into a 96-well plate (Curi Bio, ANFS- 0096) on day 9 at a density of 15,000 iGlut and 5,000 mAst per well. On day 35, the time-lapse images were acquired at ~ 5 Hz for 2 min on a Nikon A1R microscope with sCMOS camera. For quantification, peak detection was performed using the R baseline package, and HDBSCAN was used to screen out the correct clusters.
Multi-Electrode Array (MEA)An MEA assay was performed according to a previous protocol [36] with slight modification. Briefly, day- 20 iGlut were disassociated with accutase and replated with mAst into 0.1% PEI-coated 24-well MEA plate (Axion BioSystems, M384-tMEA- 24 W) at a density of 150,000 iGlut and 50,000 mAst. Neurobasal medium with 1 × Glutamax, 1 × B27, 5 µg/ml doxycycline, 10 ng/ml BDNF/GDNF/NT- 3, and 5% FBS were used on the first day after replating. In the next two days, half of the medium (300 µL) was replaced every day with fresh culture medium without FBS. Thereafter, 2/3 culture medium (no FBS) was regularly refreshed every 3 days. The culture medium was completely refreshed a day before the MEA recording. Spontaneous firing was recorded for 15 min, and the last 10 min of recording were used for data analysis. All data files were batch-processed using the Neural Metrics Tool (Axion Biosystems). For data analysis, the burst parameters were set at Poisson Surprise with a minimum surprise of 10 and Adaptive mode, Minimum number of spikes to 40, and Minimum of electrodes to 15%; Active Electrode Criterion was set at 6 spikes/min; Synchrony parameters were set at 20 ms for Synchrony Window and “none” for Synchrony Metrics.
AAV plasmid reconstruction, packaging and infectionTo express human CLU, pAAV-hSyn-hCLU-Flag was reconstructed from pAAV-hSyn-eGFP (Addgene, 58,867). First, pAAV-hSyn-eGFP was digested with BamHI and EcoRI to remove the eGFP insert. Then, the backbone DNAs (30 fmol), CLU cDNA fragment (1,400 bp; 60 fmol, synthesized from IDT, Table S15), ssDNA (single strand)-up-linker 60 bp (200 fmol, synthesized from IDT, Table S15), and ssDNA -down-linker 60 bp (200 fmol, synthesized from IDT, Table S15) were assembled using NEBuilder® HiFi DNA Assembly Master Mix (New England Biolabs, E2621S). The assembly reaction was then transformed into NEB stable competent E coli (New England Biolabs, C3040H). The transformed bacterial clone with the correct insert was determined by Sanger sequencing of plasmid DNAs, and a correct clone was expanded to prepare the plasmid DNAs for AAV packaging.
Recombinant AAVs were packaged in HEK 293 T cells (ATCC, CRL- 3216) following a previous protocol [47] with minor modifications. HEK 293 T cells were transfected with pAAV-hSyn-hCLU-Flag/pAAV-hSyn-eGFP, pUCmini-iCAP-PHP.eB (Addgene, 103,005) and pAdDeltaF6 (Addgene, 112,867) by PEI (Polysciences, 23,966) at 90% confluence. Seventy-two hours post-transfection, all cells were harvested and pelleted. AAVs were further extracted from cell pellets by AAV extraction kit (Takara, 6675) and titrated using qPCR approaches (primers are listed in Table S15).
iGlut were infected with AAV-eGFP or AAV-hCLU at 105 MOI on day 8. For calcium imaging, iGlut were infected with AAV-jRCaMP1b at 105 MOI on the next day. Between day 10 and day 12, iGlut were co-cultured with mAst or alone, following the above-described iGlut differentiation procedures. Immunostaining with Anti-Flag (Proteintech, 20,543–1-AP, Rabbit,1:200) and MAP2 antibody was used to determine the infection efficiency of AAV-hCLU on day 30 for iGlut-mAst co-culture and on day 25 for iGlut pure culture.
Lipid droplet (LD) stainingFor pure iGlut cultures, Day- 25 cells were fixed with 4% PFA, washed with PBS, and permeabilized using digitonin (Cell Signaling Technology, #16,359, ~ 0.01%) for 5 min at room temperature. The cells were then stained overnight with NeuN antibody (1:50). After the corresponding secondary antibody incubation, LipidTox (Thermo Fisher Scientific, H34476, 1:300) was applied for 2 h at room temperature. LipidTox was dissolved in PBS with 1 mg/ml DAPI. After staining, a one-time quick wash (~ 2 s) with PBS was applied before mounting the slides. The images were taken immediately after mounting on a Nikon ECLIPSE TE2000-U microscope.
For iGlut-mAst co-culture, day- 30 cells were fixed with 4% PFA, washed by PBS, and digitonin (~ 0.01%) permeabilized for 5–20 min at room temperature. Cells were then stained using antibodies against MAP2 (Synaptic System, 188,004, Guinea pig,1: 1000), GFAP (DAKO, Z0334, Rabbit, 1:500) and/or Anti-Flag (Proteintech, 20,543–1-AP, Rabbit,1:200) that were dissolved in PBS with 1% BSA. The corresponding secondary antibodies were dissolved in the same buffer to stain the cell for 2 h at room temperature. After three washes with PBS, LipidTox (1:300) or BODIPY 493/503 (Thermo Fisher Scientific, D3922, 2 µg/ml) were used to stain LDs at room temperature. The staining time was 30 min for LipidTox and 10 min for Bodipy 493/503. Both dyes were dissolved in PBS with DAPI (1 mg/ml). Bodipy 493/503 stocks were prepared in DMSO at 1 mg/ml. Cells were quickly washed (~ 2 s) once with PBS for LipidTox staining before mounting. Three quick washes (~ 2 s) with PBS were applied for BODIPY 493/503 staining [48].
Lipid transfer assayThe lipid transfer assay was performed as previously described [10, 49] with minor modifications. Briefly, following the iGlut differentiation protocol, day- 5 iGlut were replated into 24-well plates at 500,000/well and maintained until day 30. The mAst were replated into 24-well plates with Matrigel-precoated coverslips at 15,000/well and maintained for 3 days. Day- 30 iGlut were prelabeled by 2.5 μM Red C12 (BODIPY 558/568, Thermo Fisher Scientific, D3835) by adding Red C12 directly into culture medium and incubated for 18 h. iGlut were then washed twice with pre-warmed DPBS and rested for one hour in culture medium in a 37 °C incubator. mAst cells on coverslips and iGlut neurons in culture wells were washed twice with pre-warmed DPBS. Pre-warmed HBSS (Thermo Fisher Scientific, 14,175,095) containing 2 mM CaCl2 and 10 mM HEPES was added to the wells with neurons. A parafilm separator, cut to suitable size and with the center removed to form a ring, was placed above the neurons. Coverslips carrying mAst were placed face-down onto the neurons, creating a sandwich structure. The assembled cultures were then incubated at 37 °C for 4 h. After incubation, coverslips with mAst were removed, fixed and stained with Bodipy 493/503.
To confirm the lipid transfer ability of neuronal CLU within this lipid transfer system, Day- 5 iGlut (CD05, C/C) were seeded onto coverslips after replating (0.5 M/coverslip) and infected with AAV-hCLU-Flag at MOI of 105 on Day 8. From Day 26 to Day 28, these iGlut coverslips were co-cultured with mAst coverslips (prepared as described earlier in this section) using Parafilm separators. To rule out any effects of gravity, iGlut coverslips were tested in both top and bottom positions. After co-culture, all cells were fixed, permeabilized, and stained with appropriate antibodies. Flag antibody and Bodipy 493/503 were used to assess the colocalization of CLU and LDs in mAst, while MAP2 and GFAP staining were conducted to confirm cell identities (Fig. S7).
Assay for comparing CLU mRNA, intracellular, and extracellular protein levelsFor the T/T and C/C groups, Day- 10 non-proliferating iGlut cells from the CD05 line (following Ara-C treatment) were seeded either alone (0.45 M/well) or co-cultured with mAst (0.15 M/well) in 24-well plates. The cells were maintained overnight in iGlut culture medium containing 5% FBS (Neurobasal medium supplemented with B27, Glutamax, BDNF, GDNF, and NT3). The next day, the medium was carefully but completely replaced with iGlut culture medium without FBS. A separate group for pure mAst cultures was seeded at the same density and maintained in DMEM medium with 10% FBS (Fig. S6).
For hCLU overexpression groups, iGlut cells (CD05 C/C line) were first infected with AAV-eGFP or AAV-hCLU at at 105 MOI on day 8. On Day 10, the cells were replated into 24-well plates, either alone or co-cultured with mAst, following the same protocol described above (Fig. S6).
For all groups, cell mRNA, protein, and medium supernatant were collected on Day 30 according to the corresponding protocols described above.
Ketone body and lactate assayDay- 5 iGlut were directly seeded into 12-transwell (Corning, 3460, 0.4 μm pore) at 1 × 106 cells/well and cultured until day 15. mAst were seeded into the insert of 12-transwell at 200,000 cells/insert on day 13, placed into the unused wells and rested for two days in DMEM medium with 10% FBS. Then, the culture medium in the mAst insert was replaced with the iGlut culture medium (Neurobasal medium with B27, Glutamax, BDNF, GNDF, and NT3). The mAst insert was then transferred into the wells with day- 15 iGlut for 14 days of co-culture with medium change every 3 days. Then, the insert with mAst were taken out and washed three times with PBS. Next, the mAst insert was incubated in 5 times diluted DMEM medium (Thermo Fisher Scientific, 10,566,016, supplemented with Glutmax) to reach 5 mM glucose condition, without FBS and sodium pyruvate for 24 h. The supernatants were then collected at 6 and 24 h to measure the concentration of β-hydroxybutyrate (a ketone body, Promega, JE9500) and Lactate (Promega, J5021). iGlut and mAst were also collected with NP- 40 lysis buffer to determine the protein concentration using the BCA method for normalization. Data were normalized to protein concentrations of iGlut and mAst.
CellRox stainingDay- 8 iGlut and mAst were replated into 24-well plates with Matrigel-precoated coverslips at a density of 30,000/well for neurons and 10,000/well for astrocytes. iGlut/mAst were maintained in the culture medium until day 30, following the iGlut-mAst co-culture protocol described above. For CellRox staining, CellRox Deep Red (Thermo Fisher Scientific, C10422) was added to the culture medium (5 μM final concentration) and incubated for 30 min at 37 °C. After incubation, cells were fixed with 4% PFA, and processed with and without digitonin permeabilization (Fig. S8 A) to determine whether CellRox Deep Red signals can survive with this gentle permeabilization, and further stained with MAP2.
Glutamate uptake assayDay- 5 iGlut were directly seeded into 24-transwell (Corning, 3413, 0.4 μm pore) at 500,000 cells/well and cultured until day 15. mAst were seeded on the insert of 24-transwell at 40,000 cells/insert on day 13, placed into the unused wells and rested for two days in DMEM medium with 10% FBS. Then, the culture medium in the mAst insert was replaced with iGlut culture medium (Neurobasal medium with B27, Glutamax, BDNF, GNDF, and NT3). The mAst insert was then transferred into the wells with day- 15 iGlut for 14 days of co-culture with medium change every 3 days. Then, the insert with mAst were taken out, washed once with HBSS buffer (Thermo Fisher Scientific, 14,175,095) without calcium and magnesium, and incubated in the same buffer at 37 °C for 30 min. mAst insert was incubated in HBSS buffer (Thermo Fisher Scientific, 14,025,092) with 100 μM glutamate, calcium and magnesium for 3 h. The supernatants were then collected to measure glutamate concentration using the glutamate assay kit (Abcam, Ab83389). iGlut and mAst were also collected with NP- 40 lysis buffer to determine the protein concentration using the BCA method for normalization purpose. Data were normalized to protein concentrations of iGlut and mAst.
For antioxidant AD4 treatment, iGlut were co-cultured with mAst on trans-well dishes. On day 14, all medium in cultured wells and inserts was replaced with fresh medium containing AD4 (1.5 mM final concentration) for 24 h, after which the inserts with mAst were removed for glutamate uptake assay as described above.
CLU immunodepleting assayDay- 5 iGlut were replated into 12-well at 100,000 cells/well and maintained following the protocol described above. On day 27, the medium was refreshed with iGlut culture media. On day 30, the supernatants were collected and centrifuged at 3,000 rpm for 10 min at 4˚C to remove cell debris, and then frozen and stored at − 80˚C until use. CLU antibody (Santa Cruz, sc- 166907) was conjugated to magnetic beads (Thermo Fisher Scientific, 14311D) at 8 μg of antibody per mg of beads following the manufacturer’s protocol. 2 mg of antibody coupled with beads were added into 1 ml day- 30 supernatant (16 μg antibody per ml of medium) and incubated at 4 °C for 24 h. The beads were then captured on a magnetic stand, the supernatants were filtered through 0.2 μm syringe filters (Basix, 13,100,106) (CLU immunodepleted medium), and the CLU level was confirmed measured by ELISA. The conditioned medium (CM) was prepared by combining fresh DMEM medium with 10% FBS and CLU immunodepleted medium at a 1:3 ratio. For assaying the function of mAst, mAsts were seeded into 96-well plates at 40,000 cells/well 2 days before adding the CM. On day 1, the mAst culture medium was completely refreshed with CM (200 μl) and maintained until day 7 with one media change on day 4. On day 7, all CM were removed and washed with HBSS buffer (Thermo Fisher Scientific, 14,175,095) to start the glutamate uptake assay as described above. The assay data were normalized to the protein concentration of mAst from the corresponding wells.
Image quantificationNeuron morphology (MAP2 staining): All confocal z stacks (20 × objective lens) were first processed using FIJI [46] to generate maximum intensity projections. Then, neuron branches and soma were segmented by ilastik [50]. The branch length and the number of neurons (soma number) were analyzed by Cellprofiler [51] as previously described [43, 52]. The effectiveness test of neuron differentiation (MAP2 and vGlut1 staining): All confocal z stacks (40 × objective lens) were first projected with maximum intensities. The number of MAP2- and vGlut1-positive soma, as well as HuNu-positive human cells, was analyzed by CellProfiler. SYP and PSD- 95 puncta: All confocal z stacks (63 × oil immersion objective lens) were first projected with maximum intensities. ROIs of MAP2 positive dendrites were chosen uniformly from the secondary branches with ~ 50–100 μm length by FIJI [46]. The identical fluorescence intensity thresholds were applied to different images to identify SYP and PSD95 positive puncta. SYP- and PSD95- positive objects within the MAP2 mask were analyzed. For density measurements, the number of SYP or PSD95 objects was divided by the length of MAP2 (per unit was set as 10 μm) in an ROI; for area measurements, the total area of SYP and PSD95 objects were divided by the area of MAP2 in an ROI. Cellprofiler was used for all quantifications. CLU intensity: NeuN staining was used to generate a mask of soma after setting a proper intensity threshold to encircle the majority of CLU positive signals. The mean intensity of CLU signals within the NeuN mask was quantified in the 3D model of Cellprofiler. LipidTox staining in iGlut pure culture: NeuN staining was used to generate a soma mask by setting an appropriate intensity threshold to exclude signals originating from dead cells. A uniform and appropriate threshold was then applied to LipidTox signals across all groups, and the volume of LipidTox signals was calculated using the 3D model in CellProfiler. For cell counting, all confocal z-stacks (captured with a 63 × oil immersion objective lens) were first z-projected using maximum intensity in FIJI. The NeuN signals were reused to create a 2D mask, and DAPI signals within this 2D mask were selected and quantified after applying an appropriate identification in 2D model of CellProfiler. Red C12 staining in mAst: All confocal z stacks (20 × objective lens) were z projected with maximum intensity in FIJI. A uniform and proper threshold was set for Red C12 signals across different groups, and the area of Red C12 signals was calculated in Cellprofiler. The corresponding co-culture iGlut were also harvested to measure their protein concentration (by BCA kit) for normalization purposes. LipidTox staining in iGlut-mAst co-culture: All channels in confocal z stacks (20 × objective lens) were split by FIJI without any projection. Then, proper thresholds were set for both LipidTox and MAP2 signals. MAP2 staining was used to generate a 3D mask in Cellprofiler. LipidTox staining within MAP2 3D mask was identified as neuronal LDs, and non-overlapping staining was considered as astrocytic LDs. All the assayed LDs volumes were normalized by the corresponding number of cells identified by Cellprofiler. The MAP2 and GFAP volumes were also calculated after proper thresholding in the 3D model of Cellprofiler to measure the neuron-occupied areas. CellRox staining: Since all CellRox signals were located in mAst, their signal volumes were calculated without any separation in the 3D model of Cellprofiler after proper thresholding (20 × objective lens). The number of mAst was also quantified to normalize CellRox signals.
RNA-seq data analysisRaw FASTQ sequencing reads were trimmed by trim_galore and mapped to a concatenated reference genome of human (GRCh38) and mouse (GRCm39) by using Salmon (v0.11.3). All mapped transcripts (310,359, human and mouse) were further filtered based on the criterion that ≥ 75% of samples must express a transcript. Filtered human (40,162) and mouse (32,476) transcripts were analyzed for differential expression (DE) between genotypes (TT vs. CC) by using EdgeR (v4.0.16) with quasi-likelihood negative binomial generalized log-linear model. The ratio of human and mouse read counts generated from HISAT2 (v2.1.0) alignment for each sample was used as a correction factor to balance the cellular composition variance of human and mouse cells in iGlut-mAst co-culture. Counts per million (CPM)
Comments (0)