
Remember me



All reagents were used as purchased unless stated otherwise. Xylotriose and TEMPOL were bought from Megazyme and the Cayman Chemical Company, respectively. Solvents were dried in vacuo and stored over 3 Å molecular sieves. Reaction progress was monitored using thin layer chromatography (TLC) on aluminum sheets coated with silica gel 60 F254 (Merck). Detection of compounds was done using UV-absorption or by spraying with a solution of (NH4)4Ce(SO4)4⋅H2O (10 g/mL) and (NH4)6Mo7O24⋅H2O (25 g/L) in 10% sulfuric acid, followed by heating. For column chromatography, silica gel 60 M (0.04–0.063 mm) was used in combination with indicated solvents and gradient. 1H-NMR and 13C-NMR spectra were recorded on a Bruker AV-400 or Bruker AV-850 spectrometer in the given solvent. Chemical shifts are given in ppm (δ) relative to the solvent signal or tetramethylsilane (TMS) as internal standard. Given 13C-NMR spectra are all decoupled. Abbreviations used for describing signal patterns are: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet) or b (broad). High-resolution mass spectra (HR-MS) were recorded using a LTQ orbitrap (Thermo Finnigan) equipped with an electron spray ion source in positive mode (source voltage 3.5 kV, sheath gas flow 10 mL/min, capillary temperature 250 °C) with a resolution of 60,000 at m/z (400 mass range m/z = 150–2000) and dioctyl phthalate (m/z = 391.284) as lock mass. The high-resolution mass spectrometer was calibrated prior to measurements with a calibration mixture (Thermo Finnigan). Liquid chromatography mass spectrometry (LC–MS) results were recorded on a LCQ Advantage Max (Thermo Finnigan) ion-trap spectrometer (ESI +) coupled to a Surveyor HPLC system (Thermo Finnigan) equipped with a C18 column (Gemini, 4.5 mm × 50 mm, 3 µM particle size, Phenomenex) using buffers A: H2O and B: acetonitrile (ACN) or an Agilent technologies 1260 infinity LC–MS with a 6120 Quadrupole MS system equipped with buffers A: H2O, B: acetonitrile (ACN) and C: 100 mM NH4OAc. EPR spectra were recorded at room temperature with an EMX Plus EPR spectrometer (Bruker BioSpin, Germany) equipped with a SHQ resonator. EPR measurement conditions were microwave frequency: 9.88 GHz, modulation frequency: 100 kHz, modulation amplitude: 0.3 mT, time constant: 20.48 ms, power: 10.02 mW, measurement time: two min. Glass micropipettes of a volume of 50 µL (Blaubrand Intramark, Wertheim, Germany) were filled with 20 µL of sample for each measurement.
2,3,4-tri-O-acetyl-β-d-xylopyranosyl-(1 → 4)-2,3-di-O-acetyl-β-d-xylopyranosyl-(1 → 4)-1,2,3-tri-O-acetyl-d-xylopyranoside
Xylotriose (0.828 g, 2.0 mmol) was cooled to 0 °C. Acetic anhydride (2.46 mL, 26 mmol, 13 eq.) and pyridine (2.09 mL, 26 mmol, 13 eq.) were added. The reaction was stirred for 24 h at 0 °C. Remaining acetic anhydride and pyridine were removed under reduced pressure. The mixture was dissolved in EtOAc and washed three times with 1 M HCl solution, once with aqueous saturated sodium bicarb solution and once with brine. The organic phase was dried using Na2SO4, filtered and concentrated under reduced pressure. The residue was purified using silica flash column chromatography with a 10:90 → 50:50 EtOAc:n-Pent gradient (Rf = 0.27 1:1 EtOAc:n-Pent). Compound 1 was obtained as a transparent colorless oil (1.50 g, 2.0 mmol, α:β ratio of 4:5 with quantitative yield). 1H NMR (400.130 MHz, CDCl3, 293 K): δ = 6.22 (d, J = 4.0 Hz, 1H), 5.66 (d, J = 7.2 Hz, 1H), 5.10 (q, J = 7.6, 8.4 Hz, 4H), 4.99–4.94 (m, 2H), 4.88 (td, J = 4.4, 3.2 Hz, 2H), 4.78 (m, 3H), 4.58 (d, J = 6.0 Hz, 1H), 4.57 (d, J = 5.6 Hz, 1H), 4.50 (d, J = 6.8 Hz, 1H), 4.49 (d, J = 6.8 Hz, 1H), 4.10 (m, 2H), 3.98 (m, 3H), 3.82 (m, 4H), 3.67 (m, 1H), 3.50–3.30 (m, 5H), 2.10 (s, 6H), 2.05 (s, 36H). 13C NMR (100.613 MHz, CDCl3, 293 K): δ = 170.0, 169.9, 169.8, 169.7, 169.6, 169.4, 169.2, 169.0, 100.9, 100.4, 99.3, 92.3, 89.2, 75.8, 74.9, 74.3, 72.0, 71.0, 70.9, 70.3, 70.2, 70.0, 69.7, 69.4, 68.2, 63.4, 62.6, 61.5, 61.3, 21.5, 20.9, 20.8, 20.6, 20.5. HR-MS (ESI): m/z 773.211 [M + Na]+, calcd. [C31H42O21 + Na]+ 773.211.
2,3,4-tri-O-acetyl-β-d-xylopyranosyl-(1 → 4)-2,3-di-O-acetyl-β-d-xylopyranosyl-(1 → 4)-2,3-di-O-acetyl-d-xylopyranoside
Compound 1 (1.64 g, 2.2 mmol) was dissolved in THF (0.2 M, 11 mL) and 3-(dimethylamino)-1-propylamine (0.55 mL, 4.4 mmol, 2 eq.) was added. The reaction was stirred at room temperature and followed by TLC. When an undesired byproduct was detected, the reaction was diluted with DCM. The mixture was washed with 1 M HCl solution, sodium bicarb and brine. The organic phase was dried Na2SO4, filtered and concentrated under reduced pressure. Silica flash column chromatography was used for purification with a 40:60 → 70:30 EtOAc:n-Pent gradient (Rf = 0.31 50:50 EtOAc:n-Pent). Compound 2 was obtained as a transparent colorless oil (0.979 g, 1.38 mmol, 63%). 1H NMR (400.130 MHz, CDCl3, 293 K): δ = 5.44 (t, J = 8.4 Hz, 1H), 5.13–5.06 (m, 2H), 4.89 (td, J = 4.4, 2.8 Hz, 1H), 4.84–4.74 (m, 3H), 4.58 (d, J = 7.2 Hz, 1H), 4.50 (d, J = 6.0 Hz, 1H), 4.11 (dd, J = 4.8, 7.6 Hz, 1H), 4.02–3.94 (m, 1H), 3.88–3.75 (m, 3H), 3.69 (m, 1H), 3.44–3.31 (m, 2H), 2.07 (s, 21H). 13C NMR (100.613 MHz, CDCl3, 293 K): δ = 171.0, 170.4, 170.0, 169.9, 169.8, 169.5, 169.2, 100.6, 99.5, 90.4, 74.3, 71.9, 71.3, 71.1, 70.2, 69.9, 68.3, 62.4, 61.6, 20.7. HR-MS (ESI): m/z 731.201 [M + Na]+, calcd. [C29H40O20 + Na]+ 731.201.
2,3,4-tri-O-acetyl-β-d-xylopyranosyl-(1 → 4)-2,3-di-O-acetyl-β-d-xylopyranosyl-(1 → 4)-2,3-di-O-acetyl-N-phenyl-trifluoroacetimidoyl-d-xylopyranoside
Compound 2 (0.572 g, 0.81 mmol) was co-evaporated three times using distilled toluene and dissolved in dry DCM (0.2 M). While under nitrogen atmosphere, the chloro imidate reagent (0.251 g, 1.21 mmol, 1.5 eq.) and cesium carbonate (0.395 g, 1.21 mmol, 1.5 eq.) were added. The reaction was stirred at room temperature and followed by TLC. After 6 h the reaction was diluted with DCM, filtered over celite and purified by silica flash column chromatography using an EtOAc:n-Pent gradient of 10:90 → 50:50 (Rf = 0.51 50:50 EtOAc:n-Pent). Compound 3 was obtained as a white solid (0.527 g, 0.60 mmol, 74%). 1H NMR (400.130 MHz, CDCl3, 293 K): δ = 7.34–7.12 (m, 3H), 6.83 (m, 2H), 5.11 (m, 2H), 4.93–4.87 (m, 1H), 4.85–4.76 (m, 2H), 4.59 (t, J = 5.6 Hz, 1H), 4.53 (d, J = 7.2 Hz, 1H), 4.15–4.10 (m, 2H), 4.01–3.96 (m, 1H), 3.89–3.82 (m, 2H), 3.45–3.32 (m, 2H), 2.11–2.04 (m, 21H). 13C NMR (100.613 MHz, CDCl3, 293 K): δ = 170.0, 169.9, 169.8, 169.5, 169.4, 169.2, 128.9, 119.4, 100.4, 99.5, 74.3, 73.3, 72.2, 71.0, 70.2, 69.5, 68.2, 62.8, 62.0, 61.5, 20.7. HR-MS (ESI): m/z 902.230 [M + Na]+, calcd. [C29H40O20 + Na]+ 902.230.
2,3,4-tri-O-acetyl-β-d-xylopyranosyl-(1 → 4)-2,3-di-O-acetyl-β-d-xylopyranosyl-(1 → 4)-2,3-di-O-acetyl-1-TEMPOL-β-d-xylopyranoside
Compound 3 (1.553 g, 1.77 mmol) and TEMPOL (0.456 g, 2.65 mmol, 1.5 eq.) were combined and co-evaporated three times with distilled toluene. While under nitrogen atmosphere, the mixture was dissolved in dry DCM (0.2 M). The mixture was stirred for 30 min, after which it was cooled to –115 °C by using a liquid nitrogen/EtOH cooling bath. TMS-OTf (0.03 mL, 0.18 mmol, 0.1 eq.) was added in a stock solution with dry DCM after which the reaction was gradually warmed up to room temperature, while being followed by TLC. After all starting material had reacted, the reaction was quenched by adding a droplet of triethylamine. The mixture was washed with saturated aqueous sodium bicarb and brine solutions. The organic phase was dried using MgSO4, filtered, and concentrated. The resulting crude was purified using silica flash column chromatography with a 20:80 → 60:40 EtOAc:n-Pent gradient (Rf = 0.21 25:75 EtOAc:n-Pent). Compound 4 (1.461 g, 1.69 mmol, 96%) was obtained as a red colored solid. HR-MS (ESI): m/z 885.321 [M + Na]+, calcd. [C29H40O20 + Na]+ 885.324.
β-d-xylopyranosyl-(1 → 4)-β-d-xylopyranosyl-(1 → 4)-1-(1-O-4-hydroxy-3,3,5,5-tetramethylpiperidine)-β-d-xylopyranoside
Compound 4 (1.329 g, 1.5 mmol) was dissolved in MeOH (0.2 M), after which 1.99 mL 4.28 M (5 eq.) NaOMe solution was added. The reaction was stirred for 30 min at room temperature, after which quenching was performed by adding AcOH until the pH of the mixture was 7. The mixture was diluted with demi water and extracted thrice with 10 mL DCM. The aqueous phase was concentrated, and the resulting product was purified using HPLC. 0.424 g (≈50%) of material was obtained of which 8.8% was paramagnetic active as determined by EPR measurement. 0.081 g (0.12 mmol) of the obtained product was therefore dissolved in demi water (0.2 M) and treated with ascorbic acid (0.217 g, 1.20 mmol, 10 eq.). Upon full conversion to the diamagnetic compound based on LC–MS and EPR measurements, the mixture was concentrated to dryness after stirring for three hours. The obtained solid was then purified over C18 column using a H2O:ACN gradient of 100:0 → 95:5 → 90:10 → 50:50. This resulted in 22.8 mg (0.04 mmol, α:β ratio of 1:2 with 33% yield) of Dia-X3 as a white solid. 1H NMR (850.130 MHz, D2O, 293 K): δ = 4.46 (d, J = 7.7 Hz, 1H), 4.40 (d, J = 7.7 Hz, 1H), 4.38 (d, J = 8.5 Hz, 1H), 4.14–4.08 (m, 1H), 4.02 (dd, J = 6.0, 6.8 Hz, 1H), 3.99 (dd, J = 5.1, 6.8 Hz, 1H), 3.89 (dd, J = 6.0, 6.0 Hz, 1H), 3.72–3.67 (m, 3H), 3.56–3.52 (m, 1H), 3.48–3.45 (m, 2H), 3.35 (t, J = 9.4 Hz, 1H), 3.31–3.28 (m, 2H), 3.24–3.15 (m, 5H), 2.10–2.01 (bd, 2H), 1.57–1.46 (bd, 2H), 1.17 (s, 12H). 13C NMR (213.765 MHz, D2O, 293 K): δ = 102.7, 102.6, 102.4, 77.3, 77.2, 76.5, 74.7, 74.6, 73.8, 73.7, 73.6, 72.1, 66.1, 63.9, 63.8. HR-MS (ESI): m/z 570.275 [M + H]+, calcd. [C24H43NO14 + H]+ 570.275.
β-d-xylopyranosyl-(1 → 4)-β-d-xylopyranosyl-(1 → 4)-1-TEMPOL-β-d-xylopyranoside
Dia-X3 (6.5 mg, 11.0 µmol) was dissolved in a minimal amount of demi water (22 µL, 0.5 M). MnO2 (4.8 mg, 55.2 µmol, 5 eq.) was added and the reaction was stirred for 40 min. After full conversion was observed by LC–MS, the mixture was filtered thrice using PTFE/L 0.45 µm filters. Remaining MnO2 was removed using a centrifuge and carefully pipetting the supernatant, which contained Para-X3. After concentrating the sample 4.4 mg (7.70 µmol, 70%) of Para-X3 was obtained as a brown-colored solid. At least 90% of the produced Para-X3 was in the paramagnetic state, based on MS, NMR and EPR data (Figs. S1–S3). HR-MS (ESI): m/z 568.260 [M]+, calcd. [C24H42NO14]+ 568.260.
Site-directed mutagenesis and protein productionSite-directed mutagenesis, protein production and purification were performed as described (Saberi et al. 2024).
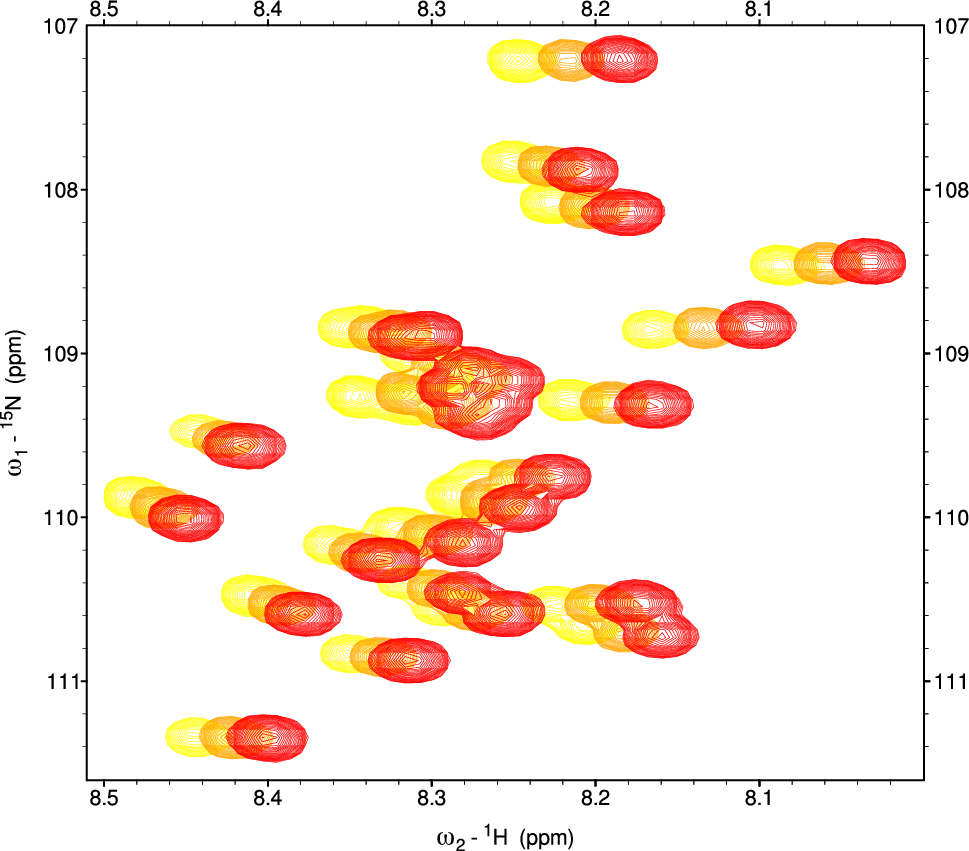
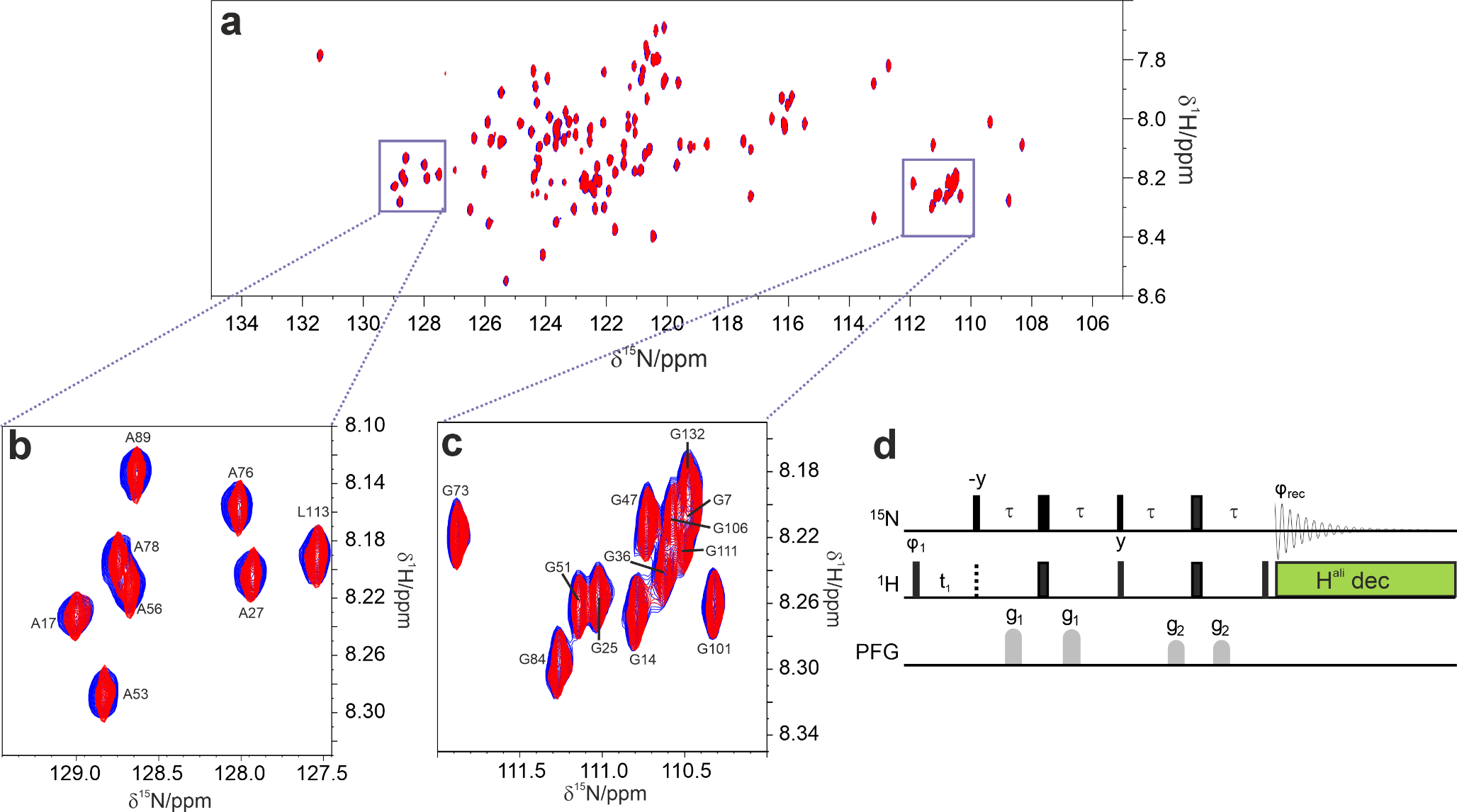
Protein NMR spectroscopySolutions of 15N-labeled BcX E78Q and BcX E78Q-Y69A (100 µM) were titrated with Dia-X3 and Para-X3. Concentrations of up to 50 mM for Dia-X3 and 8 mM for Para-X3 were used. Samples were prepared separately, and each contained 25 mM sodium acetate buffer (pH 5.8) and 10% D2O for lock. 1H − 15N HSQC spectra were obtained on a Bruker HD Avance 850 MHz spectrometer with a cryoprobe, at 20 °C, using 3 mm tubes. In one experiment Para-X3 was reduced in the NMR tube with ascorbic acid to study the effect of reduction in a single sample. The assignment, processing, and analysis of the HSQC spectra were conducted as detailed before (Saberi et al. 2024).
Spectra were processed with Topspin (Bruker) and analyzed using CCPN Analysis v.2.4.2 (Vranken et al. 2005). The weighted average chemical shift perturbations (CSP) for backbone amides (Δδavg) was calculated according to Eq. 1 (modified after Williamson 2013):
$$\Delta \delta_ = \sqrt + \frac}^ }$$
(1)
where δH and δN are the CSPs for 1H and 15N nuclei. Titration data were fitted to a 1:1 model Eq. (2), which simplifies to (3) when the ligand concentration [Lt] is much higher than the protein concentration [Pt] (Arai et al. 2012; Wang et al. 1996; Williamson 2013):
$$ \Delta \delta _} = \Delta \delta _} \left\ + [P_ ] + [L_ ]} \right) - \sqrt + [P_ ] + [L_ ]} \right)^ - 4[P_ ]} [L_ ]} \right\}/2[P_ ] $$
(2)
$$_}=\frac}_}]}}_}+}_}]}_}$$
(3)
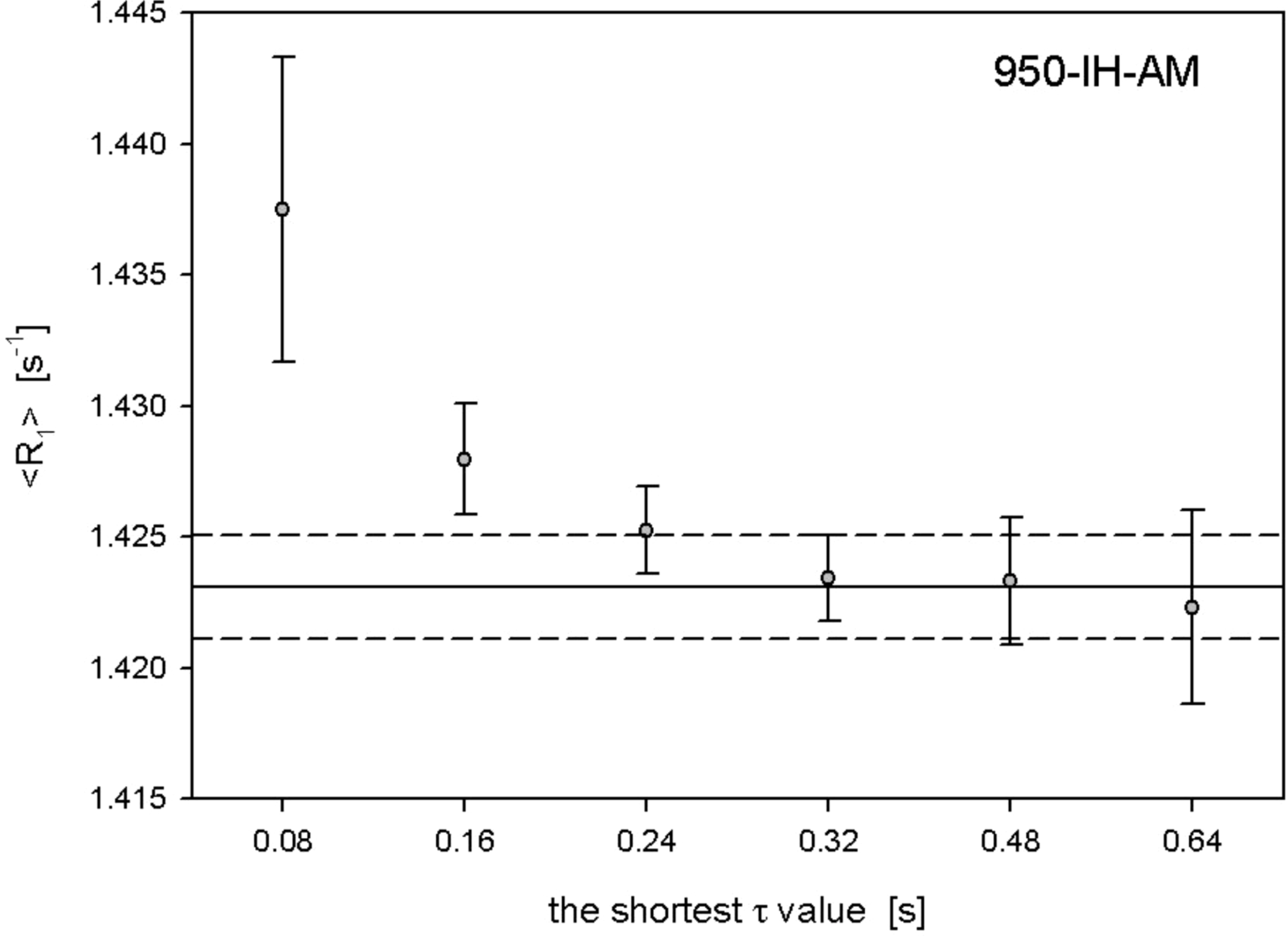
where Δδobs and Δδmax are the observed and maximal change in the chemical shift and KD is the dissociation constant. The error in KD was estimated by manually adjusting KD to higher and lower values and observing the range over which the fit of the CSP data to the model remained within the peak picking error bars of the CSPs.
The fractional populations of the free and bound states were calculated using the peak height (I) as follows:
$$f_ = \frac }} + I_ }},\,f_ = \frac }} + I_ }}$$
(4)
Here, ffree and fbound represent the fractions of the protein in the free and bound states. The uncertainties in the peak heights represent the noise level in the spectra. The dissociation constant KD was determined using:
$$_=\frac_.\left[_\right]}_}$$
(5)
In this equation, [Lfree] represents the concentration of the free ligand, which was approximated as the total ligand concentration [Ltotal] because the ligand was in large excess relative to the protein. The mean KD value was determined by averaging KD values across the used residues.
Comments (0)