
Remember me






All 30 healthy adult male sprague dawley (SD) rats (male, 8–10-week-old, 250–280 g) were purchased from the Animal Experiment Center of Dalian Medical University. They were grouped and given a period of time to adapt to the laboratory environment. All animal experiments were approved by the Ethics Committee of the Animal Experiment Center of Dalian Medical University and performed following the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (Ethical No. AEE23127, Feb. 28th 2023). They were housed in a specific-pathogen-free animal central laboratory with 50% relative humidity, 22 ± 2 °C temperature, and a 12-h light and dark cycle. They were free to consume standard rat food and water. The animals were excluded if the animal died prematurely, preventing the collection of behavioral and histological data. At the end of the observation period, the rats were humanely euthanized with carbon dioxide in accordance with the institutional guidelines. All animal experiments were reported in line with the ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments).
Cell culture and treatmentMouse hippocampal neuronsMouse hippocampal neurons (HT22; RRID: CVCL_0321) were provided by the Laboratory of Dalian Medical University. Considering that neurons are more susceptible to OS and have higher metabolic demands, HT22 cells were chosen as the in vitro research subject in this study. HT22 cells were cultured in dulbecco's modified eagle medium (DMEM) high-glucose medium (MeilunBio, MA0560, Dalian, China), fetal bovine serum, penicillin and streptomycin. HT22 cells were cultured in an incubator at 37 °C with 5% CO2. They were subcultured at 3-day intervals and carried out using passages 4–8. An OS-induced cell injury model was established by treating HT22 cells with H2O2 (400 μM, Sigma-aldrich, HX0640, Darmstadt, Germany) for 6 h in vitro. And the "Mdivi-1" group comprises HT22 cells exposed to 400 μM H2O2 while also being treated with the mitophagy inhibitor 10 μM mdivi-1 (MedChemExpress, HY015886, New Jersey, USA) in vitro. The concentrations of H2O2 and mdivi-1 were referenced from previously published literature [28, 37] and it has been confirmed in this study that the essence of this model is apoptosis caused by OS and inhibition of the mitophagy pathway.
hNSCsThis study was approved by the Clinical Research Ethics Committee of the First Affiliated Hospital of Dalian Medical University (Ethical No. LCKY2016-60, Dec. 2nd 2016). hNSCs were isolated from the human fetal forebrain tissue of healthy pregnant women at 10–12 weeks of gestation who requested induced abortion, all of whom provided informed consent. Our research group has independently established a seed bank for NSCs. And the tests for human immunodeficiency virus, syphilis, hepatitis B, and other pathogenic microorganisms were negative, as we previously reported [38]. The extraction method of hNSCs was as described earlier. Within 6 h after miscarriage, the forebrain tissue isolated from the aborted fetus was transported by cold chain to our research group's stem cell preparation center, which has been registered with the China Food and Drug Administration. hNSCs were extracted strictly according to the established protocol in our laboratory. hNSC basal medium (Stemcell Technologies, 05751, Vancouver, Canada), epidermal growth factor (Stemcell Technologies, 78,136.1 l, Vancouver, Canada), basic fibroblast growth factor (Stemcell Technologies, 78,134.1, Vancouver, Canada), heparin (Stemcell Technologies, 07980, Vancouver, Canada), penicillin and streptomycin (Solarbio, P1400, Beijing, China) were used to culture the qualified hNSCs. hNSCs were cultured in adherent flasks coated with matrigel (Corning, 356,234, New York, USA). Cells were cultured in an incubator (Escolifesciences, IFC-240, Singapore) at 37 °C with 5% CO2. The hNSCs were subcultured at 3-day intervals and carried out using passages 4–8. We strictly controlled the quality of hNSCs during passage by monitoring cell viability and marker expression.
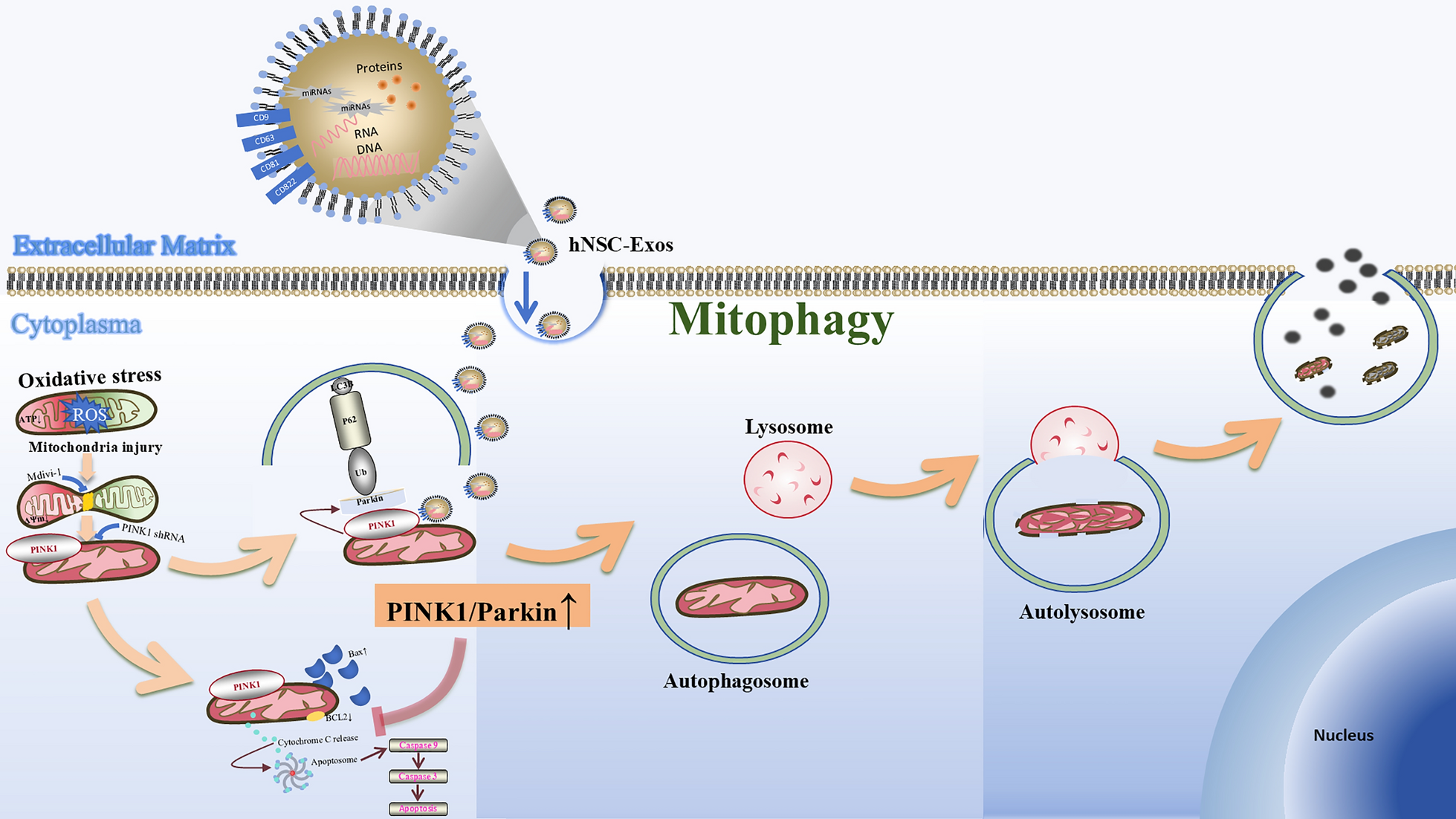
hNSC-Exos isolationThe ultracentrifugation was employed to purify the exosomes. hNSCs were cultured for 3-day by exo-free FBS cell culture medium, the cell supernatant was collected and transferred to a centrifuge tube (Sparkjade, GD0001, Jinan, China). Repeated gradient centrifugation was performed at 200 g for 10 min, 2,000 g for 20 min, and 20,000 g for 30 min. The supernatant was collected, filtered through a 0.22-μm sterile filter (Merck, SLGPR33RB, California, USA), and added to an ultracentrifuge (Beckman, Optima XPN-100, Brea, USA). After centrifugation at 100,000 g and 4 °C for 1.5 h, the precipitate was resuspended in a little sterile phosphate buffered saline (PBS) (MeilunBio, MA0015, Dalian, China) and quantified in aliquots. The obtained-exosomes were quantified using bicinchoninic acid reaction kit (Thermo Fisher Scientific, 23,227, Massachusetts, USA). In this study, the density of hNSC-Exos was 1–1.2 g/ml, and they were divided into 40 μg/tube according to experimental requirements. Before storage, quality control was carried out by detecting particle size through Nanoparticle tracking analysis (NTA) and protein integrity through western blotting (WB). Once the control standards were met, the samples were quickly frozen at -80 ℃ to avoid multiple thawing cycles. In vitro, hNSC-Exos precipitates were resuspended in fresh medium at 40 μg/ml and experiments were performed at 20 μg/group. hNSC-Exos were co-cultured with OS-induced HT22 for 6 h. In vivo, hNSC-Exos precipitates were resuspended in 7 μg/μL of hNSC-Exos dissolved in 10 μL of PBS. The therapeutic concentration of hNSC-Exos selected in this study was based previous literature published by our research group [39].
NTAhNSC-Exos were diluted 40 times with sterile PBS and then filtered with 0.22 μm sterile filter. The diameter and potential of exosomes were measured by ZetaView (Particle Metrix, PMX-120-Z, Munich, Germany). Data were analyzed using the ZetaView software 8.02.31. The particle size is mostly concentrated in 50–150 nm, with a main peak of about 80–120 nm and a negative potential.
Transmission electron microscopy (TEM)TEM (Philips, TECNAI 20, Eindhoven, Netherlands) was used to observe the ultramorphology of the hNSC-Exos. After cleaning, the hNSC-Exos were fixed with 2% osmotic acid solution, dehydrated with a gradient of ethanol, mixed with acetone, immersed in an embedding plate for polymerization, and finally stained with uranyl acetate solution for observation. The TEM parameters are set to an acceleration voltage of 80–100 kV, an objective aperture of 20–30 μm, and an exposure time of 0.5–1 s. The complete hNSC-Exos are cup-shaped or double concave disc-shaped.
In addition, TEM was used to observe the mitochondrial morphology and autophagy structure of neurons in vitro. In vitro, hNSC-Exos were co-cultured with OS-induced HT22 for 6 h. After collecting HT22 cells, the above operation method was repeated. After fixation and dehydration treatment, the mitochondria were embedded and sliced to expose them in the observation field, and finally stained. The TEM parameters are set to an acceleration voltage of 80–120 kV, an objective aperture of 30–50 m, and an exposure time of 0.5–2 s. Normal mitochondria are elliptical or elongated in shape, with a diameter of about 0.5–1 μm and a length of 1–10 μm. They have a bilayer membrane structure and tightly arranged mitochondrial cristae. And the autophagosome is clearly visible as two membrane bilayers separated by electron translucent pores.
WBProteins were extracted from the cell, hNSC-Exos and animal models in each group using radio-immunoprecipitation assay lysis buffer (Thermo Fisher Scientific, 89,901, Massachusetts, USA) and quantified using a BCA kit (Thermo Fisher Scientific, 23,227, Massachusetts, USA). The protein samples were all normalized to the same concentration. Samples were added to a Bis–Tris preformed gel (Meilun, MA0445, Dalian, China) for electrophoresis using electrophoresis equipment (Life Technologies, EI0001, California, USA) set at 150 V and 600 mA for 1 h. Polyvinylidene fluoride membranes (Merck, IPFL00010, California, USA) were used for membrane transfer. A transfer solution (Meilun, MA0121, Dalian, China) was used, and the transfer conditions were set at 600 V and 300 mA for 1 h. Subsequently, they were blocked with skim milk (BD Difco, 232,100, Colombia, USA) and incubated overnight with primary antibodies, including CD9, CD63, CD81, TSG101, Calnexin, Bax, BCL2, Caspase 3, Cleaved caspase 3, Beclin, LC3B, Parkin, PINK1 and GAPDH. The next day, the membranes were then incubated at room temperature for 2 h with second antibodies. The bands were made visible through the use of an ECL kit (Thermo Fisher Scientific, 34,580, Massachusetts, USA) and chemiluminescence instrument (SuperSignal West Atto Supersensitive substrate, Thermo Fisher Scientific, A38554, Massachusetts, USA) were used for exposure development. Relative protein expression was normalized to that of GAPDH. ImageJ software was used to quantify the protein expression levels. The typical molecular weight range of hNSC-Exos marker proteins ranges from 25 to 95 kDa and the detailed information of all antibodies used is summarized in Table 1.
Table 1 All antibodies and primer sequences for qPCR used in the manuscripthNSC-Exos uptakeTo determine the endocytosis of exosomes by hNSC-Exos which were initially labelled with the red fluorescent dye PKH26 (Sigma aldrich, MKCL4676, Darmstadt, Germany) concerning the manufacturer's instructions. In detail, after fluorescent labeling of hNSC-Exos with PKH26, hNSC-Exos rinsed with PBS were added to diluter C, and the mixture of PKH26 and diluter C was added to mix thoroughly. The reaction was terminated through serum addition, centrifuged (12,000 g, 4 °C, and 15 min), and washed several times with PBS before use. In vitro, PKH26-hNSC-Exos were co-cultured with HT22 for 6 h, and the cells were fixed, permeabilized, and incubated with actin-tracker green-488 (Beyotime, C2201S, Shanghai, China) for fluorescence staining of neuronal microfilaments. Subsequently, they were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Solarbio, C0065, Beijing, China) and observed under a laser confocal microscope (Yokogawa, CV1000, Tokyo, Japan). In vivo, PKH26-hNSC-Exos were stereotaxically injected into the brain of middle cerebral artery occlusion (MCAO) rats during ischemia–reperfusion. After 24 h, the rats were sacrificed, and the brain tissues were frozen (Detailed operation approaches will be discussed later). Finally, the cells were stained with DAPI and observed under a laser confocal microscope.
Flow cytometryCarboxyfluorescein diacetate, succinimidyl ester (CFDA SE, Beyotime, C1031, Shanghai, China) was used to label the internalized hNSC-Exos. CFDA SE was prepared as a 5 μM working solution in anhydrous DMSO and mixed with hNSC-Exos, followed by incubation at room temperature. The labeling reaction was terminated by adding complete medium. The CFDA SE-labeled hNSC-Exos were co-cultured with HT22 cells for 6 h. Afterward, the cells were collected, resuspended in PBS, and analyzed by flow cytometry (Sony, SH800, Tokyo, Japan). If hNSC-Exos were not taken up by the cells, they were non-fluorescent. By comparing the cell sorting of hNSC-Exos that were co-cultured with cells to those that were not, we can determine whether hNSC-Exos had been internalized by the cells.
HT22 cell apoptosis was detected using a fluorescein isothiocyanate-Annexin V/PI apoptosis kit (Thermo Fisher Scientific, BMS500FI, Massachusetts, USA). We had set up three groups in vitro experiments, respectively. The "Control" group represents untreated HT22 cells, the "H2O2" group comprises HT22 cells exposed to 400 μM H2O2, and the "hNSC-Exos" group consists of HT22 cells exposed to 400 μM H2O2, followed by treatment with 20 μg hNSC-Exos. We ensured that each batch of cells in vitro was highly similar and randomly grouped, and this standard was maintained in subsequent experiments. After preparing the HT22 cell suspension, a binding buffer was added, followed by the addition of fluorescent dye-coupled annexin V. The cells were incubated at 20 °C-25 °C for several minutes. A binding buffer was readded, and a propyl iodide staining solution was added. The cells were sorted using flow cytometry.
Cell viability assaysCell viability of neurons was analyzed based on the inability of trypan blue (Thermo Fisher Scientific, T10282, Vancouver, USA) dye to penetrate the normal intact cell membrane of living cells. As described above, HT22 cells were treated. Cells were stained by trypan blue after treatment. Stained HT22 cells were considered non-viable, and unstained HT22 cells were considered viable. Cell survival rate = (Total cell count-Stained cell count) / Total cell count × 100%. Damaged cell rate = (Total cell count-Unstained cell count) / Total cell count × 100%. Statistical analysis of the three groups was performed to assess the cell survival and death rates of each group. Among them, the normal HT22 cells in the control group were not treated and were used as the negative control group.
In addition, a lactate dehydrogenase (LDH) release was used to indicate cytotoxicity and evaluated using an LDH cytotoxicity detection kit (DOJINDO, CK12, Tabaru, Japan). As described above, HT22 cells were treated. Cells were plated into a 96-well plate and various groups were treated with specific modeling techniques. Subsequently, 100 µL of the reaction mixture supplied with the kit was added to each well. The plate was then incubated under cell culture conditions at 37 °C with 5% CO2 for 30 min. The optical density value at 490 nm using a microplate reader (DeTie, HBS-1096A, Nanjing, China). Statistical analysis of the three groups was performed to assess the cytotoxicity of each group (ImageJ, 1.52).
TdT-mediated dUTP nick end labeling (TUNEL) stainingThe TUNEL apoptosis kit (Beyotime, C1090, Shanghai, China) was used to detect neuronal apoptosis in vitro and in vivo. Briefly, neuronal different groups in vitro and MCAO rat brain slices (frozen section manipulation methods are detailed later) were incubated with anti-NeuN antibodies (1:100, Abcam, Ab177487, Massachusetts, USA), and then incubated with a mixture of biotinylated dUTP catalyzed by dinitrotoluene and goat anti-rabbit 488 (1:500, Abcam, Ab150077, Massachusetts, USA) in the dark. An unlabeled solution was used as a negative control. Images were captured and photographed using a fluorescence microscope. The ratios of TUNEL-positive cells were calculated separately for randomly selected fields in different groups and images of representative areas by ImageJ.
Quantitative real time PCR (qRT-PCR)qRT-PCR was performed to assess the gene expression in the experimental groups. Total RNA was extracted from the cell samples using the RNA extraction kit (QIAGEN, 21,017, Dusseldorf, Germany). The concentration and purity of RNA were assessed using a NanoDrop ND-1000 spectrophotometer (ThermoFisher Scientific, Waltham, USA). After extracting RNA, the reverse transcription kit (Accurate Biology, AG11728, Beijing, China) was utilized to perform reverse transcription, converting RNA into cDNA. qRT-PCR was performed on a real-time PCR machine (Bio-Rad, 1,855,200, California, USA) using a SYBR Green Pro Taq HS Premixed qPCR kit (Accurate Biology, AG11746, Beijing, China). Relative RNA expression levels were assessed using the 2−△△Ct method. All the tests were performed in triplicate and β-actin was used as a genomic DNA control. qRT-PCR primers were purchased from Sangon Biotech (Shanghai, China) and the primer sequences are summarized in Table 1.
Immunofluorescence stainingHT22 cells and sections were fixed with 4% paraformaldehyde, permeabilized (Beyotime, China, P0096), blocked (Beyotime, P0260, Shanghai, China), and incubated with primary antibodies overnight, including Nestin, SOX2, Bax, BCL2, PINK1, Beclin, LC3B. The next day, they were treated with second antibodies and incubated for 1 h. DAPI was used to mark the nuclei. Images were captured by confocal microscopy using the same imaging threshold and exposure time for each experiment. The number of positive cells per field was calculated with ImageJ software. The detailed information of all antibodies used is summarized in Table 1.
Mitochondrial membrane potential and apoptosis detectionWe used mitochondrial membrane potential and apoptosis detection kit with Mito-Tracker Red CMXRos and Annexin V-FITC (Beyotime, C1071, Shanghai, China) to detect mitochondrial membrane potential and apoptosis of HT22 cells. As described above, HT22 cells were treated. Following the manufacturer's instructions, the fluorescence was observed by fluorescence microscopy and analyzed by ImageJ. Red fluorescence marks viable cells that retain mitochondrial membrane potential, whereas green fluorescence marks cells that have undergone apoptosis or necrosis.
Intracellular ROS detectionThe ROS assay kit-highly sensitive DCFH-DA (Dojindo, R252, Tabaru, Japan) was used to detect ROS production. After removing the medium, a high-sensitivity DCFH-DA dye working solution was added after washing with Hank's Balanced Salt Solution (HBSS) (Beyotime, C0218, Shanghai, China). As described above, HT22 cells were treated. HT22 cells were cultured in an incubator for 30 min, rewashed with HBSS, and observed using fluorescence microscopy. The relative fluorescence intensity of DCFH was analyzed by ImageJ.
Mitochondrial reactive oxygen species (MtROS) detectionThe MitoSOX Red probe (MedChemExpress, HY-D1055, New Jersey, USA) was used to detect mtROS production. As described above, HT22 cells were treated. After removing the medium, a MitoSOX Red working solution was added after washing with HBSS. HT22 cells were cultured in an incubator for 30 min, rewashed with HBSS, and observed using fluorescence microscopy. The relative fluorescence intensity was analyzed by ImageJ.
Malondialdehyde (MDA) detectionMDA detection kit (Beyotime, S0131S, Shanghai, China) was used to detect lipid oxidation levels. As described above, HT22 cells were treated. After treatment, the cells were washed with PBS and lysed. After centrifugation, the supernatant was collected for MDA detection. Following the manufacturer's instructions for MDA, the absorbance of cellular MDA at 532 nm was measured using an enzyme-labeled instrument, and the molar concentration of MDA was calculated based on the standard curve.
Monodansylcadaverine (MDC) stainingAutophagy Staining Assay Kit with MDC (Beyotime, C3018, Shanghai, China) was used to rapidly detect autophagy in HT22 cells. As described above, HT22 cells were treated. After the medium was removed, MDC staining solution was added and incubated for 1 h at 37 °C in the dark. When autophagy occurred, the green fluorescence was observed by fluorescence microscopy and analyzed by ImageJ.
RNA sequencing (RNA-seq)To evaluate the changes in gene expression between control group, H2O2 group and hNSC-Exos group, total RNA was extracted from HT22 cells using standard extraction methods (Accurate Biology, AG21024, Beijing, China). After quality checks for purity, concentration, and integrity, oligo dT magnetic beads were specifically bound to the poly (A) tail of the mRNA to remove other RNAs, and the purified mRNA was fragmented. The treated mRNA was used as a template for reverse transcription to synthesize cDNA and for PCR amplification with primers. After purification, PCR was performed to establish a library. After quality inspection, the library was sequenced on a sequencer according to the manufacturer's operating guidelines. The R package was employed to analyze differences in mRNA expression levels between the two groups. DEGs with a P-value < 0.05 and FDR < 0.05 were considered statistically significant. The mRNA exhibiting differential expression was subsequently subjected to enrichment analysis based on Gene Set Enrichment Analysis (GSEA).
Double-label adenovirus mCherry-GFP-LC3B transfection and autophagy assayHT22 cells were inoculated in confocal dishes (NEST, 726,001, Wuxi, China), and the experiment was conducted according to the transfection procedure for double-label adenovirus mCherry-GFP-LC3B (Beyotime, C3011, Shanghai, China). Upon infection with the Ad-mCherry-GFP-LC3B adenovirus, the mCherry-GFP-LC3B fusion protein is observed to emit a uniform yellow fluorescence across the cytoplasm under fluorescence microscopy in the absence of autophagy, resulting from the merged signals of mCherry and GFP. In contrast, during autophagic conditions, the protein localizes to the autophagosomal membranes, where it forms distinct yellow puncta. Following fusion of the autophagosomes with lysosomes, the fluorescence transitions to red puncta, a shift attributed to the selective quenching of GFP fluorescence. The number of yellow spots representing autophagic bodies and red spots representing autophagic lysosomes were counted.
Construction of HT22-KO-PINK1 Stable strainHT22 cells were infected with PINK1-knockout lentivirus (Beyotime, L16306, Shanghai, China), and a stable KO-HT22 strain was constructed using Clustered Regularly Interspaced Short Palindromic Repeats -associated protein 9 (CRISPR/Cas9) technology. HT22 cells with good growth and 80% cell density were selected, spread on 6-well plates, and cultured in an incubator to determine the optimal multiplicity of lentivirus infection. The concentrated viral particles were added to the HT22 cell suspension. After 24 h of transfection, the medium containing the virus was discarded and replaced with a new medium for further culturing, and puromycin was added to screen for stable cell strains. Approximately 72 h after infection, different groups of KO-PINK1 were set up, and the expression of PINK1 in the protein was detected to evaluate the transfection efficiency, and the group with higher transfection efficiency was selected for subsequent experiments.
Animal modelAccording to the experiment requirement, male SD rats were divided into three groups, namely sham group, MCAO group, and hNSC-Exos group (10 rats/group). All rats were anesthetized through intraperitoneal injection of 2.5% tribromoethanol (MedChemExpress, T48402, New Jersey, USA). They were fixed on the operating table in the supine position, and the neck skin was cut with surgical scissors to expose the carotid triangle. Similarly, the rats in the sham group were exposed only to the blood vessels, and the wounds were sutured immediately without further treatment. Rats in the MCAO and hNSC-Exos groups were further operated on. The right common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were blunted separately to protect the vagus nerve. The proximal ends of the common and ECAs were ligated, and the ICA was clipped using an arterial clamp. An incision was made at the distal end of the CCA using ophthalmic scissors, and a thread plug (RWD, MSRC37B200PK50, Shenzhen, China, 250-280G, 5–6 mm) was slowly inserted into the ICA until it reached the bifurcation of the middle cerebral artery. After occlusion of the blood flow for 1.5 h, the thread plug was removed to restore blood flow for reperfusion was restored to successfully simulate CIRI [40]. Within 30 min after restoring blood flow, PBS (10 μL, sham group) or hNSC-Exos (7 μg/μL of hNSC-Exos dissolved in 10 μL of PBS, hNSC-Exos group) were delivered through an automatic brain stereo stereoscope (RWD, 71,001-S, Shenzhen, China), respectively, at the anterior (1.3 mm), lateral (3.5 mm), and ventral (6.5 mm) sites. The rats in the Sham group underwent the same operation of middle cerebral artery ischemia–reperfusion injury as the rats in the MCAO model group. It differs from the hNSC-Exos group, in which the Sham group was given PBS treatment to exclude the effect of the surgical procedure itself on the experimental results. We referred to the fourth edition of the Rat Brain Stereotaxic Coordinates. During the process, we adopted a double-blind test, that is, the subsequent examiners did not know the experimental rats in each group to minimize possible errors owing to subjective factors. Animals were excluded if they died prematurely. The legend shows the number of nude rats that provided data for analysis in each experiment.
Behavioral testsThe behavior tests were conducted by independent researchers who were unaware of the experimental group, and the data was analyzed by a separate researcher. The monitoring time points of neural function were referred to the published literature [41, 42].
Modified neurological severity score (mNSS)The severity of nervous system damage in SD rats was evaluated. The mNSS scores were assessed before surgery, and 1, 2, 3, 5, 7 days after surgery, including motor (muscle strength status and abnormal movement), sensory (vision, touch, and proprioception), reaction, and balance tests. The total scores ranged from 0 to 18, with lower scores indicating more robust function, and higher scores indicating more severe impairment.
Cylinder experimentEvaluation of motor sensory screening and detection of tactile responses and asymmetry in SD rats were conducted. Rats should familiarize themselves with the testing environment before the experiment. Before surgery, and 1, 3, 5, and 7 days postoperatively, each rat was placed in a transparent resin glass cylinder with a diameter of 20 cm and a height of 40 cm. The spontaneous standing exploration activity was observed for 5 min, and a video was recorded. The videos were analyzed to assess the preference of the left and right forelimbs of the rats for exploration. The asymmetry index was calculated as follows: percentage of the ipsilateral forelimb alone lying on the wall-percentage of the control forelimb alone lying on the wall. The greater the asymmetry index, the worse the motor coordination and the greater the motor cortex damage.
Adhesive tape removal experimentEvaluation of motor sensory screening and detection of tactile responses and asymmetry in SD rats were performed. Before starting the experiment, the two front paws of the rats were pasted on the dorsal side of the rats’ toes with a circular paper with a diameter of 8 mm (the animal was not covered, and were shaved before the experiment) for at least 3 days. The time for removing labels from both forelimbs of the rats will be recorded, and training will be conducted at least four times a day to select rats that can remove labels quickly from both forelimbs for inclusion. Before surgery, and 1, 3, 5, and 7 days postoperatively, a 5-mm diameter circular adhesive label was attached to the relatively hairless wrists of the rats to observe and record the time of label removal on the injured side.
Triphenyltetrazolium ammonium chloride (TTC) stainingAll rats were randomly selected for TTC staining. After the rats were sacrificed (24 h later), the intact brain tissue was removed and placed in a refrigerator at −80 °C for 10 min. Subsequently, the brain tissue was cut into slices of approximately 2 mm thickness and incubated in 2, 3, 5-triphenyltetrazolium ammonium chloride (Solarbio, T8170, Beijing, China) for 30 min at 37 °C in the dark, with stirring to maintain uniform staining. After staining, a 4% paraformaldehyde (Biosharp, BL539A, Hefei, China) solution was fixed and photographed. Areas of normal brain tissue appeared red, whereas infarcted areas appeared pale. ImageJ software was used to calculate the relative infarct volume of the brain tissue (percentage of cerebral infarct area/ total brain area).
Frozen sectionIt is prefered that the tissues to be examined using frozen sections be made of fresh material. The harvested rat brain tissues were fixed with 4% paraformaldehyde for 24 h. Subsequently, brain tissues were dehydrated using gradient concentrations of 10%, 20%, and 30% sucralose-PBS solutions. After dehydration, the brain tissue was soaked with opti-mum cutting temperature compound (Leica, 39,475,237, Heidelberg, Germany), followed by tissue fixation with a freezing microtome and serial slicing to a thickness of 20 μm.
Statistical analysisAll data were statistically analyzed using GraphPad Prism 9.0. The raw data of in vitro and in vivo experiments (such as protein expression levels, fluorescence intensity, etc.) were standardized using internal references. In addition, intention-to-treat analysis was used for statistical analysis of animal studies. For excluded experimental rats, we substituted their last available data to minimize the interpretation of the final findings due to animal exclusion. All experiments were repeated at least three times. Results are expressed as mean ± standard deviation. The student’s t-test was utilized to analyze differences between two groups. For experiments with more than two groups, one-way analysis of variance (ANOVA) was applied, followed by Tukey's post hoc test and Student–Newman–Keuls (SNK)/ Least Significant Difference comparisons (LSD). P ≤ 0.05 was considered statistically significant.
Comments (0)