Cell line and culture
Human iPSC control line SCTi003-A (StemCell™ Technologies #200–0511, Vancouver, BC, Canada), a hiPSC line derived from peripheral blood mononuclear cells of a healthy female donor, was used in this work. iPS cells were cultured in 60 mm tissue culture dishes coated with Matrigel (hESC-qualified matrix, Corning, New York, NY, USA), using mTESR™ Plus medium (StemCell™ Technologies) at 37 °C with 5% CO2 and 20% O2, with medium changes every other day. The iPSCs were passaged every four days using ReleSR™ (StemCell™ Technologies) or StemPro Accutase (Gibco, Waltham, MA, USA) and 10 µM Rock inhibitor (StemCell™ Technologies) at a 1:3–1:5 splitting ratio.
CRISPR-Cas9-mediated gene KO
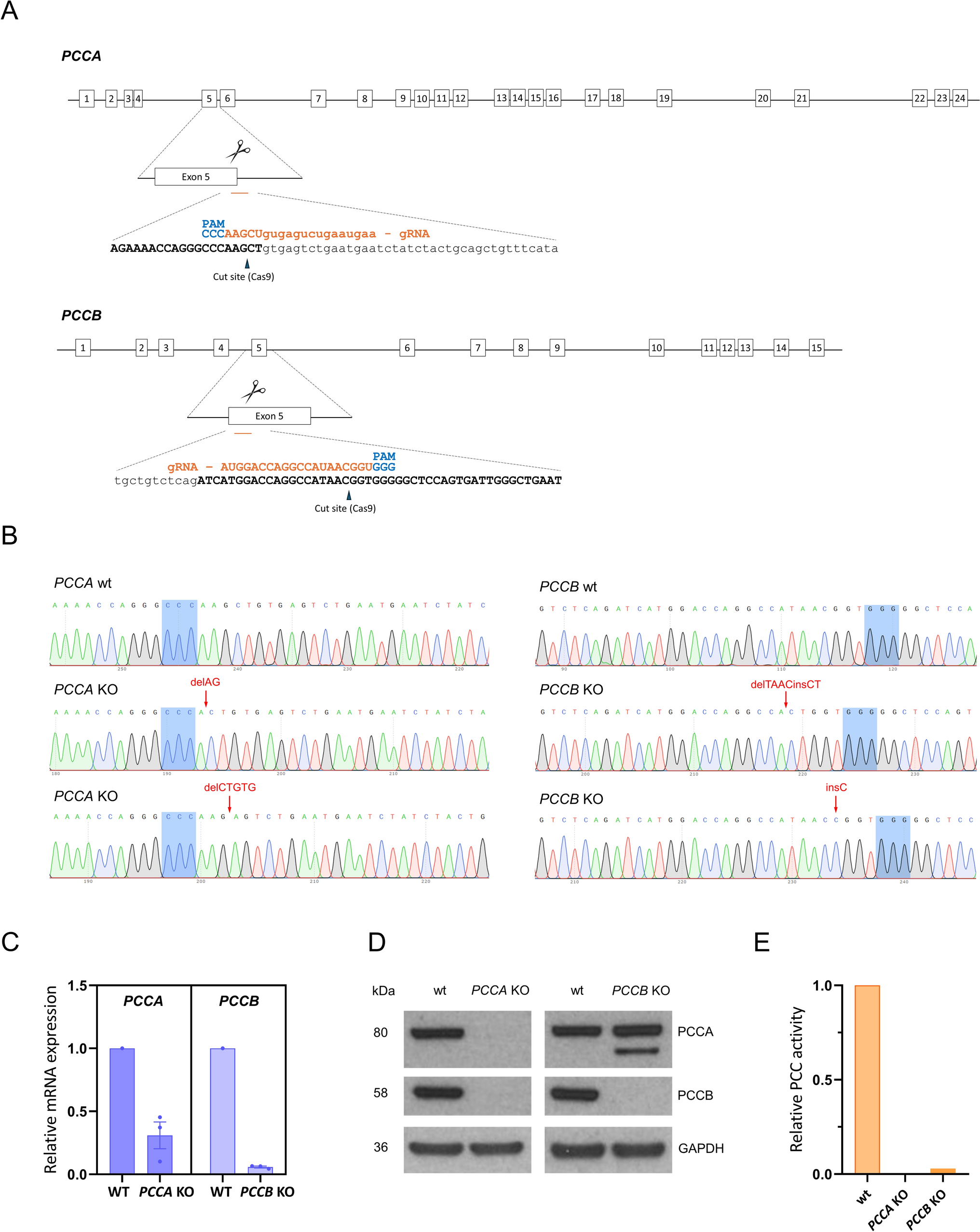
The online tools available at https://bioinfogp.cnb.csic.es/tools/breakingcas/ and http://crispor.gi.ucsc.edu/ were used to design guides with high specificity for PCCA and PCCB target sites while minimizing predicted off-targets located in genomic regions (Table 1). On the day of nucleofection, 2 × 105 cells (80% confluency and passage 4) were nucleofected using a Neon Electroporation Kit (Invitrogen, #MPK1025, Waltham, MA, USA) and the Neon Transfection System (Invitrogen), according to provider´s instructions. Ribonucleoprotein (RNP) complexes were formed by the combination of 45 pmol of Cas9 (Streptococcus pyogenes, Integrated DNA Technologies (IDT), Newark, NJ, USA) and 55 pmol of RNA duplex constituted by tracrRNA and RNA guide (IDT); and with 55 pmol of enhancer (IDT). Cells were transferred into a single well of a Matrigel-coated 12-well plate and cultured in mTESR™ Plus medium. Several days after nucleofection, the cells were transferred to Matrigel-coated 60 mm dishes in mTESR™ Plus medium. Cells were then sorted using a FACSAria Fusion (BD Biosciences, Franklin Lakes, NJ, USA) into Matrigel-coated 96-well plates with mTESR™ Plus medium supplemented with 1X CloneR (StemCell™ Technologies) for clonal expansion.
Table 1 Primers and RNA guides used in the studyPCC enzymatic assay
PCC activity was assayed by measuring the enzyme-dependent incorporation of radiolabelled bicarbonate into non-volatile products, as previously described [9].
Karyotype analysis
Cells were exposed to 10 μg/ml Colcemid® Solution (Irvine Scientific, Santa Ana, CA, USA) for 3 h at 37 °C, then dissociated using accutase, treated with a hypotonic solution, and fixed with Carnoy’s fixative in preparation for karyotype analysis. A minimum of 20 metaphase cells were analyzed. Karyotype analysis was performed by the Molecular Cytogenetics and Genome Editing Unit from Centro Nacional de Investigaciones Oncológicas (CNIO, Madrid, Spain).
DNA extraction and PCR mismatch
Genomic DNA was extracted from iPSCs using QIAamp DNA Mini Kit (Qiagen, Venlo, The Netherlands) following the manufacturer´s recommendations. Editing was verified by PCR mismatch using 100 ng of DNA from the cell population, the specific primers for target knockout analysis (Table 1), and the Supreme NZYTaq II 2 × Green Master Mix (NZYTech, Lisbon, Portugal).
Sequencing
PCR products were cloned in pGEM®-T Easy Vector System I (Promega Corporation, Madison, WI, USA). Plasmid DNA was purified using NZYMiniprep Kit (NZYtech) and was subjected to Sanger sequencing (Macrogen, Seoul, South Korea). At least 20 colonies were sequenced. Deep sequencing was performed using amplicons generated in a first PCR with fusion primers containing a specific sequence plus a common tag (Table 1). Libraries preparation and sequencing were carried out at Fundación Parque Científico de Madrid (Campus Cantoblanco, Madrid, Spain) under protocols developed and optimized for next-generation amplicon sequencing. For off-target analysis, PCR products were purified using E.Z.N.A Cycle Pure kit (Omega Bio-Tek, Norcross, GA, USA) before Sanger sequencing.
RNA extraction, RT-PCR and RT-qPCR
To assess the splicing effects in the PCCA KO cell line, total RNA was isolated using TRIzol® Reagent (Life Technologies, Waltham, MA, USA), and 1 μg of RNA was retrotranscribed using NZY First-Strand cDNA Synthesis kit (NZY Tech). RNA from the unedited iPSC line was used as a control. PCR was performed using the primers hybridizing to exons 2 and 7 of the PCCA gene (Table 1), and FastStart Taq DNA polymerase (Roche Diagnostics GmbH, Mannheim, Germany). PCR products corresponding to transcripts were separated on an agarose gel and purified using a QIAquick gel extraction kit (Qiagen) before Sanger sequencing.
To assess the effect of gene KO on their respective target mRNA levels, cDNA was obtained by retrotranscription of 500 ng of total RNA as described above. PCCA and PCCB were then amplified with specific primers designed upstream of the edition site (Table 1), using PerfeCTa SYBR Green FastMix (Quanta Biosciences, Beverly, MA, USA) in a CFX Opus 384 real-time PCR system (Bio-Rad, Hercules, CA, USA) following the manufacturer’s instructions. Data were analysed using Bio-Rad CFX Maestro software, and relative quantification to the wild-type iPSC line was performed according to 2−∆∆CT method, using GAPDH as an endogenous control.
Immunoblot analysis
Protein extracts were prepared from frozen cell pellets through a freeze–thaw lysis process using a buffer containing Tris–HCl pH 7.4, 10% glycerol, 150 mM NaCl, 0.1% Triton X-100, and protease and phosphatase inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA). Lysates were then centrifuged for 10 min at 4 °C, after which the supernatant was collected, and protein concentration was measured using the Bradford method (Bio-Rad). 40 µg of protein from each sample were loaded onto 4–12% NuPAGE™ Precast Gels (Invitrogen). After electrophoresis, proteins were transferred to a nitrocellulose membrane in an iBlot Gel transfer device (Invitrogen), then incubated with the primary antibodies at 4 °C overnight (Table 2), and subsequently incubated with the secondary antibodies at room temperature for 1 h (Table 2). GAPDH was used as a loading control. Enhanced chemiluminescence reagent (ECL, GE Healthcare, Chicago, IL, USA) and SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, Waltham, MA, USA) were used for protein detection.
Table 2 Antibodies used in the studyImmunostaining
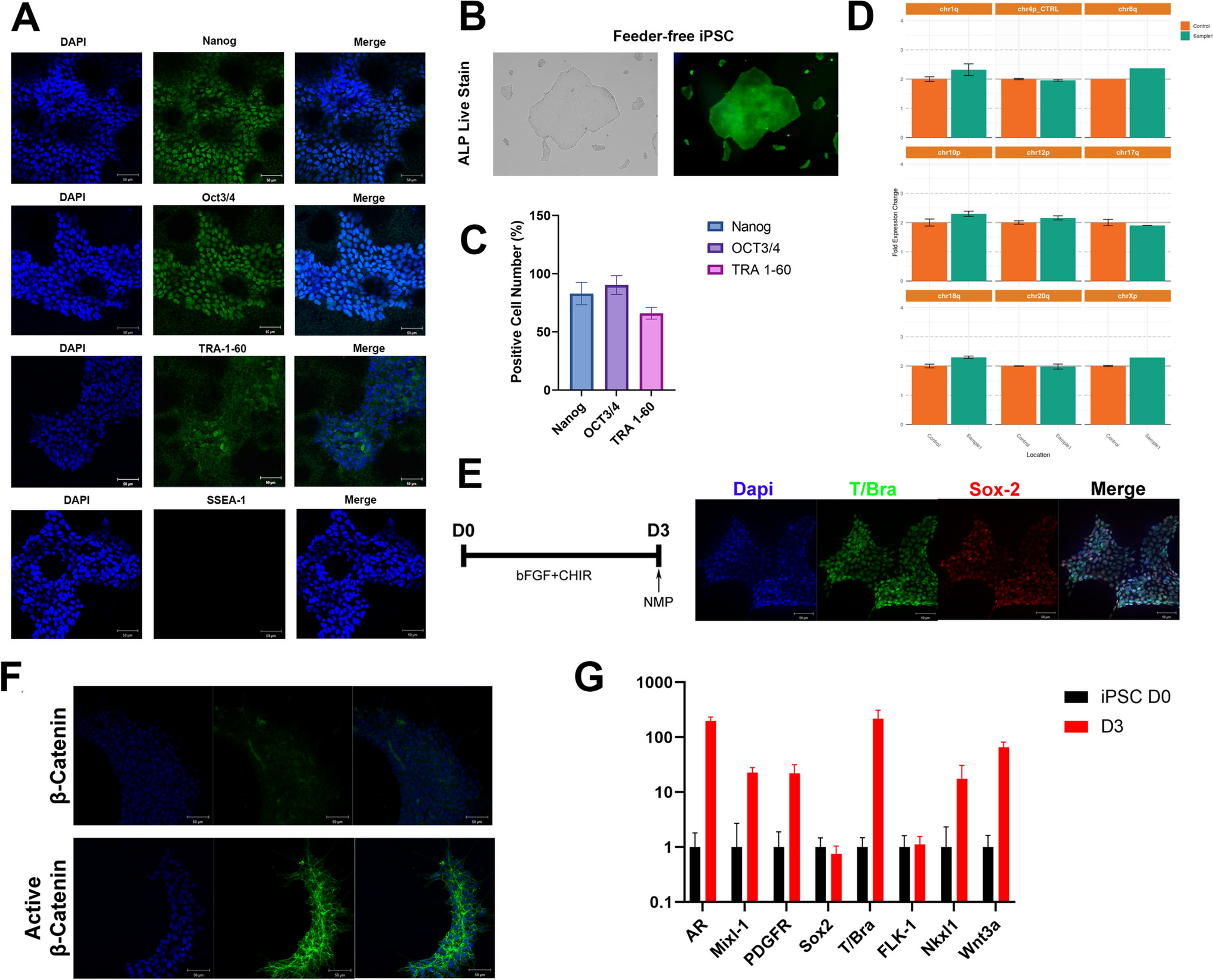
iPSCs were seeded onto matrigel-coated 15 μ-Slide 8 well culture plates (Ibidi, GmbH, Gräfelfing, Germany), fixed with Formaline Solution 10% (Sigma-Aldrich), and stained with anti-OCT4/NANOG/SOX2/SSEA-3/SSEA-4/TRA-1–60 and TRA-1–81 at 4 °C overnight using the dilutions previously described [10]. Tris-buffered saline was used for washing, blocking and antibody incubation solutions supplemented with Triton-X100 (Sigma-Aldrich) (Nuclear markers: 0.1% for washes and 0.3% for blocking and antibody incubation solutions) and Tween-20 (Merck Millipore, Burlington, MA, USA) (Surface markers: 0.2% for washes and 0.4% for blocking and antibody incubation solutions). Blocking and antibody incubation solutions were also supplemented with 3% Donkey Serum (Sigma-Aldrich). After several washes, cells were labelled with secondary antibodies for 2 h at 37 °C, followed by 30 min at room temperature. For nucleus staining DAPI (Invitrogen, 1:10,000) was used. Images were obtained using a Nikon A1R fluorescence microscope. Antibodies details are in Table 2.
Flow cytometry analysis
The pluripotency markers SSEA-4, TRA-1–60, and TRA-1–81 were also examined by flow cytometry, as detailed in [10], using a BD FACSCanto™ A instrument (BD Biosciences), FACSDiva and FlowJo v10.10 software program. Unstained iPSCs and matching isotype antibodies served as negative controls to eliminate non-specific fluorescence data.
In vitro three-germ-layer differentiation assay
Embryoid body (EB) formation was applied by plating the dissociated cells onto 96-well v-bottom, low attachment plates (Deltalab, Barcelona, Spain). Emerging EB were replated on matrigel-coated 15 µ-Slide 8 well culture plates (Ibidi) for another 18 days with different mediums as described [10]. Cells were fixed with Formaline Solution 10% and stained with endodermal (α−1-Fetoprotein, AFP), mesodermal (α-Smooth muscle actin, SMA) and ectodermal (β-III-Tubulin Tuj1, TUJ1) differentiation markers (Table 2).
Short tandem repeat (STR) analysis
DNA fingerprinting analysis was conducted using the AmpFLSTR® Identifiler® PCR Amplification Kit (Thermo Fisher Scientific). For this process, 1 ng of DNA was used to evaluate highly polymorphic regions containing short tandem repeats by amplifying the following markers: D7S820, CSF1P0, Th01, D13S317, D16S539, vWA, TPOX, D5S818, and Amelogenin for determining sex. Samples were analyzed on a 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA), and data processing was done using GeneMapper® v4.0 software at the Parque Científico de Madrid, Campus Moncloa, UCM, Madrid, Spain.
Mycoplasma detection
Mycoplasma test was performed using the PCR method [11]. A positive sample with mycoplasma was used as a control.






Comments (0)