Ethics statements
Stem cell transplantation was approved by the Institutional Ethics Committee of Xijing Hospital, The Fourth Military Medical University. Written informed consent was obtained from the participants. The animal study protocol was approved by the Animal Welfare and Ethics Committee of the Fourth Military Medical University and was performed according to the 'Guidelines for the Care and Use of Laboratory Animals'.
Subjects
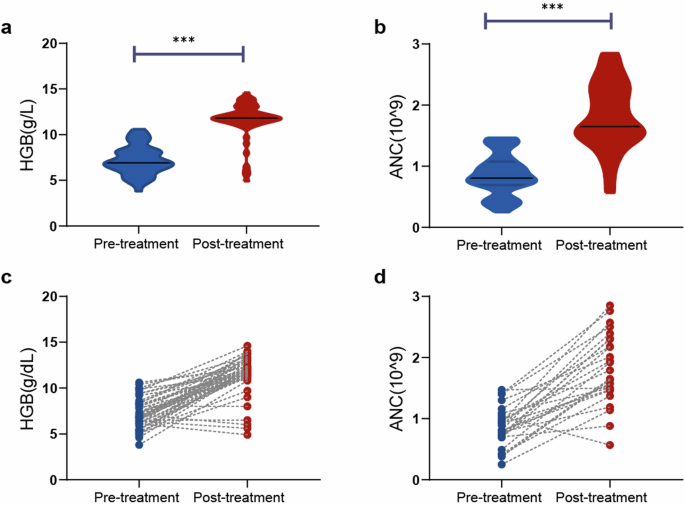
Patients with decompensated cirrhosis who received stem cell transplantation were included in the analysis (NCT01728688). Decompensated cirrhosis was diagnosed on the basis of ultrasound or computerized tomography, upper digestive tract endoscopy, and patient history. Chronic hepatitis B was diagnosed on the basis of patient history, serologic tests, and biochemical tests. All patients received standard medical treatment, which consisted of antiviral therapy and the management of complications such as ascites, variceal bleeding, hepatorenal syndromes, and hepatoencephalopathy, according to updated guidelines.62,63,64 In the transplantation group, eligible patients were randomly assigned to receive stem cell treatment. During infusion, the collected cells were transplanted into the recipient's liver via the hepatic artery. Follow-up examinations were routinely performed after treatment. These patients were divided into responsive (n = 27) and nonresponsive groups (n = 31) according to improvements in the MELD score (MELD change ≤ −10%) at 6 months after treatment.
Animals
Fstl1+/− mice were generated as previously described,65 and the mice were backcrossed onto the C57BL/6J background for at least 12 generations before use. Six- to eight-week-old male C57BL/6J mice were purchased from Beijing Vitong Lihua Laboratory Animal Technology Co., Ltd. (Beijing, China). All animals were housed and cared for in a pathogen-free airflow cabinet and allowed free access to food and water.
Liver fibrosis models
Liver fibrosis was induced by the intraperitoneal (i.p.) injection of 7 ml/kg CCl4 (20% solution in olive oil) twice per week. Mice were treated with mouse bone marrow-derived MSCs (1 × 106) for early efficacy evaluation at the indicated times. At the designated time points, the mice were euthanized with phenobarbital sodium by i.p. injection, and the livers were harvested for further analyses. Mice with liver fibrosis were treated with IgG or 22B6 (50 µg/each, i.v.), CsA (Selleck, 15 mg/kg, i.p.) or the CCR2 inhibitor PF4136409 (Selleck, final dose of 20 µg each, i.p.) before MSC treatment. Fstl1+/− mice with liver fibrosis were treated with FSTL1 (2 µg each, i.v.) or FSTL1 (R&D systems, 2 µg each, i.v.) + PF4136409 (final dose of 20 µg each, i.p.) before MSC treatment.
Flow cytometry
Hepatic mononuclear cells were isolated from the liver with 30% Percoll (GE Healthcare) according to the established method without collagenase digestion.66 Single-cell suspensions were first incubated with anti-mouse FcR blocking reagent (BioLegend) and then labelled with the mixed fluorochrome-conjugated antibodies (BioLegend) PerCP/Cyanine5.5 anti-mouse CD45.2, PE/Cyanine 7 anti-mouse F4/80, FITC anti-mouse/human CD11b, APC/Cyanine 7 anti-mouse CX3CR1 and PE anti-mouse Ly6C, APC-conjugated anti-mouse CCR2, APC-conjugated anti-mouse CD45.1, PE-conjugated anti-mouse F4/80, PerCP/Cyanine5.5-conjugated anti-mouse Ly6C, Brilliant Violet 510-conjugated anti-mouse Ly-6G and the Zombie Aqua Fixable Viability Kit (BioLegend). Flow cytometry was conducted with a FACS Canto II flow cytometry system (BD Biosciences). All the data were analysed with FlowJo software (FlowJo LLC, version 10.0.7). Liver macrophages or the Ly6C–CX3CR1+ subset were sorted from viable CD45+F4/80+CD11b+Ly6G–hepatic mononuclear cells with a FACSAria III flow cytometry system (BD Biosciences) for further analysis. Circulating monocyte analysis was performed by staining whole blood after the erythrocytes were lysed.
Single-cell RNA sequencing (scRNA-seq)
CD45+ hepatic mononuclear cells were sorted and the cell suspension was loaded into Chromium microfluidic chips with 3’ (v2 or v3, depending on the project) chemistry and barcoded with a 10× Chromium Controller (10X Genomics). RNA from the barcoded cells was subsequently reverse-transcribed, and sequencing libraries were constructed with reagents from a Chromium Single Cell 3’ v2 reagent kit (10X Genomics) according to the manufacturer’s instructions. Sequencing was performed with an Illumina platform (Nova6000, Novegene, Tianjin, China) according to the manufacturer’s instructions (Illumina)
Single-cell transcriptome analysis
We use fastp to perform basic statistics on the quality of the raw reads. The raw read sequences produced by the Illumina pipeline in FASTQ format were subsequently preprocessed via Trimmomatic software to obtain clean reads. For the generation and analysis of single-cell transcriptomes, the raw reads were demultiplexed and mapped to the reference genome via the 10X Genomics Cell Ranger pipeline (https://support.10xgenomics.com/single-cell-geneexpression/software/pipelines/latest/what-is-cell-ranger) via default parameters. All downstream single-cell analyses were performed via Cell Ranger and Seurat (Macosko et al., 2015; Satija et al., 2015) unless otherwise mentioned. In brief, for each gene and each cell barcode (filtered by CellRanger), unique molecule identifiers were counted to construct digital expression matrices. Secondary filtration by Seurat was performed as follows: a gene expressed in more than 3 cells was considered expressed, and each cell was required to have at least 200 expressed genes. Some of the foreign cells were filtered out. The Seurat package was used to normalize the data and perform dimensionality reduction, clustering and differential expression. We used the Seurat alignment method for canonical correlation analysis (CCA) [Nat. Biotechnol. 36, 411–420 (2018).] for the integrated analysis of datasets. For clustering, highly variable genes were selected and the principal components based on those genes were used to construct a graph, which was segmented with a resolution of 0.6.
Cell culture
Mouse bone marrow-derived MSCs was generated from bone marrow from the tibias and femurs of 6- to 10-week-old mice as previously reported.67 The cells were cultured in DMEM supplemented with 10% heat-inactivated FBS, 2 mM glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin (Invitrogen). The cells were used before the 5th passage. Mouse MSCs were used for all MSC therapy mouse models. The characteristics of the MSCs were determined via flow cytometry. The antibodies (anti-CD29, anti-CD44, anti-CD140a, and anti-sca-1) conjugated to PE were purchased from BioLegend (Supplementary Fig. 10a). Osteoblast, adipocyte, and chondrogenic differentiation media were purchased from Cyagen Biosciences (HUXUC-90021, HUXUC-90031, and HUXUC-9004) and used according to the manufacturer’s instructions (Supplementary Fig. 10b). Intracellular lipid or calcium deposits were stained with Oil Red O or Alizarin Red S. The presence of proteoglycans, which indicate chondrogenic differentiation, was verified by toluidine blue staining after 21 d of pellet induction in a 15 mL tube. THP-1, a human leukaemia monocytic cell line, was purchased from ATCC and maintained in RPMI 1640 GlutaMAX medium supplemented with 10% (v/v) heat-inactivated foetal bovine serum (FBS) and a 1% (v/v) mixture of penicillin and streptomycin (all reagents from Thermo Fisher Scientific) in a humidified incubator containing 5% CO2 at 37 °C. BMDMs were also generated from the bone marrow of the tibias and femurs of 6- to 10-week-old mice. We obtained nonadherent cells after 12 h of adherent culture and suspended them in BMDM culture medium (DMEM supplemented with 5% fetal bovine serum, and 50 ng/mL M-CSF) at a concentration of 107 cells/mL. Then, the BMDMs were cocultured with or without FSTL1 (100 ng/ml) for 48 h. Chemical inhibitors, Cycloheximide (CHX, 10 μg/ml, Selleck), Baf-A1 (0.5 nM, Selleck) and bortezomib (10 nM, Selleck), were used for inhibiting protein synthesis, blocking lysosome degradation or NF-κB signal transduction.
Mouse MNCs were generated from bone marrow from the tibias and femurs of 14- to 16-week-old mice with established liver fibrosis as previously reported.10 Whole bone marrow was first isolated by flushing the dissected femurs and tibiae of mice with liver fibrosis. The suspension was then centrifuged at 400 × g for 10 min at 4 °C, resuspended in 3 mL of sterile saline, and layered on top of 4 mL of Ficoll Paque Plus (GE Healthcare). The cells were then centrifuged at 2500 × g for 30 min at 4 °C in a swinging bucket rotor centrifuge without brakes. MNCs were isolated by removing of the resulting thin mononuclear cell layer (second layer from the top). Total MNCs were counted with a haemocytometer, washed twice with sterile saline, and resuspended in sterile saline at a final concentration of 1.0 × 106 cells/ml.
Adoptive transfer of CD45.1+ bone marrow cells
We injected CCl4-induced CD45.2+ recipient mice (including WT and Fstl1+/–, IgG, FSTL1 and FSTL1 + PF4136309) with 5 ×106 bone marrow cells. Then, we analyzed the subsequent distribution of CD45.2–CD45.1+ donor-derived tissue-resident macrophages (F4/80+CD11b+) 48 h after bone marrow cell transfer.
RNA sequencing (RNA-seq) analysis
Total RNA was isolated via TRIzol (Invitrogen) according to the manufacturer’s instructions. RNA sequencing libraries were generated with insert sizes ranging from 370 to 420 bp and sequenced via the Illumina HiSeq 2500 platform (Gene Denovo-Guangzhou, China). The image data measured by the high-throughput sequencer were converted into sequence data (reads) by CASAVA base recognition. Raw data (raw reads) of fastq format were first processed through in-house Perl scripts. In this step, clean data (clean reads) were obtained by removing reads containing adaptors, reads containing N bases and low-quality reads from the raw data. Moreover, the Q20, Q30 and GC contents the clean data were calculated. All the downstream analyses were based on the clean data. Each sample produced an average of 6.0 G of data. The clean reads were mapped to the reference genome with HISAT2 (v2.0.5) software. Data processing and analysis were performed with the R programming language. For quantification of gene expression levels, featureCounts (v1.5.0-p3) was used to count the number of reads mapped to each gene. The expected number of fragments per kilobase of transcript sequence per million base pairs sequenced (FPKM) value of each gene was subsequently calculated on the basis of the length of the gene and the number of reads mapped to that gene. Differential expression analysis of two conditions/groups (two biological replicates per condition) was performed with the DESeq2 R package (1.20.0). DESeq2 provides statistical routines for determining differential expression in digital gene expression data with a model based on the negative binomial distribution. The resulting P-values were adjusted with the Benjamini and Hochberg’s approach for controlling the false discovery rate. padj < =0.05 and |log2(foldchange)| >= 1 were set as the thresholds for significantly differential expression. Differentially expressed genes (DEGs) were included for further functional analysis based on Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. The details of all the identified genes are listed in Supplementary Table 2.
Blood biochemistry measurements
Mouse serum was obtained at each time point, and the levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST) and albumin (ALB) were determined with an automatic biochemistry analyser (Hitachi 7600-120, Hitachi, Japan).
Hydroxyproline assessment
Hydroxyproline (HYP) levels were assessed with a kit (Jiancheng Bioengineering Institute, Nanjing) following the manufacturer’s protocol. Equal amounts (in weight) of liver tissue samples were also analysed with the kit.
Cytokine measurements
The human serum concentrations of cytokines (FSTL1, TNF-α, IL-1β and IL-6) were determined using a commercially available Human Magnetic Luminex Assay (R&D Systems) in accordance with the kit-specific protocols provided by the manufacturer. Plates were read on a Bio-Plex 200 system (Bio-Rad Laboratories, Hercules, CA, USA) and analyzed using Bio-Plex Manager software (Bio-Rad Laboratories) with a five-parameter model used to calculate final concentrations and values (expressed in pg/ml). Reference samples were run on each plate to evaluated assay consistency, and all samples were run in a blinded manner. The serum concentrations of cytokines in CCl4-induced liver fibrosis model were determined using ELISA kits (FSTL1, Raybiotech; TNF-α, IL-1β and IL-6, Neobioscience).
Cell fractionation
The cell homogenates were fractionated on Percoll gradients essentially as previously described. Briefly, the cells were rinsed twice with PBS supplemented with 2 mM EDTA and 5 mM EGTA, resuspended in ice-cold homogenization buffer (HB), which consisted of 10 mM HEPES (pH = 7.5), 0.25 M sucrose, 1 mM EDTA, 0.2 mM phenylmethylsulfonyl fluoride (PMSF), and 1 M leupeptin, and homogenized with 22 strokes of a Dounce homogenizer. The homogenate was diluted with an equal volume of fresh HB and centrifuged at 400 × g for 10 min at 4 °C to precipitate unbroken cells and nuclei. Postnuclear supernatants were adjusted to a final concentration of 27% Percoll–0.25 M sucrose using a 90% Percoll stock solution and were then layered over a 0.5 ml sucrose cushion consisting of 10x HB. The gradients were centrifuged for 90 min at 25,000 × g in a fixed-angle rotor without braking. A total of 15 fractions were collected manually, starting from the top of the gradient, and 1–11 fractions were collected for analysis via western blotting.
NanoBRET assays
HEK293 cells were seeded into 6-well plates (8 × 105 cells per well) and transfected with FuGENE HD transfection reagent (Promega) when the cells reached ~70% confluency. The cells were transfected with a BRET donor (Promega, 200 ng of CCR2-Nanoluc per well) along with 2 µg of BRET acceptor per well (Promega, for example, HaloTag-Rab4, HaloTag-Rab11 or HaloTag-Rab7). After 20 h, the transfected HEK293 cells were replated into 96-well plates, the cells were divided into two groups, and HaloTag NanoBRET 618 Ligand or DMSO vehicle was added. The samples were treated with or without FSTL1 (100 ng/ml) for at least 16 h. Live-cell kinetic detection was performed via the NanoBRET Nano-Glo® Kinetic Detection System, and a 1x solution of Nano-Glo Vivazine substrate (a 1:100 dilution of the stock reagent) was prepared in Opti-MEM® I Reduced Serum Medium, with no phenol red + 4% FBS substrate. Vivazine solution was added to each well, and the plate was incubated for 30–60 min at 37 °C and 5% CO2 to equilibrate the substrate luminescence. CCL2 (100 ng/ml) was added and kinetic measurements of donor emission (460 nm) and acceptor emission (618 nm) were collected every 3 ~ 5 min via a NanoBRET PPI Assay-compatible luminometer (Promega Biotech, Beijing, China). BRET measurements were taken ~5 min apart for 60 min.
Statistical analysis
The quantitative data are presented as the mean ± standard error of the mean (SEM). For comparisons between two groups, a two-tailed Student’s t-test was used; when the data were not normally distributed, a nonparametric Mann–Whitney U-test was used. For comparisons across multiple groups, one-way analysis of variance (ANOVA) with Tukey multiple comparison test was used.
Comparisons between the same individual were performed with Wilcoxon’s matched-pairs test. The relationship between two variables was evaluated with the Spearman rank correlation test. P-values of < 0.05 were considered statistically significant. The 'pROC' package was used to plot the ROC curves. The optimum cut-off point was determined by the greatest Youden index, and the sensitivity and specificity were calculated on the basis of the cut-off point. All the statistical analyses were performed with GraphPad Prism 8.0 (GraphPad Software, CA), SPSS software version 21.0 (SPSS Inc.) and R (version 3.6.1).








Comments (0)