2.1 Materials and methods
All starting materials and solvents required for the synthesis were purchased from Sigma-Aldrich, TCI or Carlo Erba company, and used as supplied without any further purification. Hydrogel HydroMed™ D4 (ether-based hydrophilic urethane) was obtained from AdvanSource BiomaterialsCorp. pH buffers were prepared by dissolving citric acid and sodium phosphate in MilliQ water to get buffers at pH 2.6, 3.0, 3.6, 4.0, 4.6, 5.0, 5.6, 6.0, 6.6, 7.0, 7.6 and 8.0 (see table s1 in supplementary information for the details). Thin-layer chromatography (TLC) was used for monitoring the reaction outcome using silica gel aluminum plates ALUGRAM®Xtra SIL G/UV254 from Macherey–Nagel.Column chromatography has been conducted with a Armen Instrument Spot™ Flash II Liquid Chromatography using premade silica column (RediSep®Rf from Teledyne Isco or PuriFlash® from Interchim). Microwave irradiation were performed using Monowave 300 from Anton Paar 1H NMR and 13C NMR spectra were recorded on JEOL-FT NMR 400 MHz equipped with a large band probe 40TH5AT/FG and an automatized tuning module. Deuterated solvents used are specified for each spectra and have been produced by Euriso-top®. The signals corresponding to the residual protons of the deuterated solvent were used as an internal reference. All chemical shifts (δ) are quoted in ppm, and coupling constants J are expressed in Hz. The following abbreviations are quoted to describe spin multiplicity in 1H NMR: s = singlet, d = doublet, t = triplet, q = quartet, qu = quintuplet, dd = doublet of doublets, m = multiplet. HRMS spectra have been made by the mass spectrometry service from the Institut de Chimie des Substances Naturelles (ICSN), at the research center from Gif sur Yvette (Centre de Recherche de Gif—http://www.icsn.cnrs-gif.fr), or from the Institut de Chimie Moléculaire et des Matériaux d’Orsay.
2.2 Synthesis and characterization of compounds
tert-Butyl piperazine-1-carboxylate 1 [
13]
The synthesis has been conducted following the procedure reported by Gobbo et al. [13] with piperazine (1.00 g, 11,6 mmol, 1 eq) and Boc2O (1.27 g, 5.85 mmol, 0.5 eq), adding 1.5 eq of NEt3 and using ethyl acetate (AcOEt) for the extraction. The pure product is obtained as white crystals (718 mg, yield = 66%). 1H-NMR (CDCl3, 400 MHz): 1.46 (s, 9H); 2.81 (t, J = 5 Hz, 4H); 3.39 (t, J = 5 Hz, 4H).
tert-Butyl 4-(4-hydroxybenzyl)piperazine-1-carboxylate 2 [
14]
tert-Butyl piperazine-1-carboxylate (718 mg, 3.85 mmol, 1 eq), 4-hydroxybenzaldehyde (471 mg, 3.85 mmol, 1 eq), AcOH (0.07 mL, 1,15 mmol, 0.3 eq) and NaBH(OAc)3 (1.23 g, 5.78 mmol, 1.5 eq) are dissolved in 15 mL of 1,2-dichloroethane and placed under argon. After 72h, the mixture is then evaporated, dissolved in H2O and extracted with AcOEt. The organic layer is dried with MgSO4, filtrated and evaporated. The white solid obtained is adsorbed on SiO2 and purified by column chromatography (eluent: AcOEt). The pure product is obtained as a colorless oil which crystallizes after 3 days at RT as a colorless solid (842 mg, yield = 75%). 1H-NMR (CDCl3, 400 MHz): 1.45 (s, 9H); 2.39 (t, J = 5 Hz, 4H); 3.43 (t, J = 5.2 Hz, 4H); 3.45 (s, 2H); 6.75 (d, J = 8.4 Hz, 2H); 7.5 (d, J = 8.4 Hz, 2H).
Ethyl 6-(tosyloxy)hexanoate 3 [
15,
16]
The synthesis has been conducted following the procedure reported by Burns et al., with ethyl 6-hydroxyhexanoate (1.2 mL, 7.38 mmol, 1 eq) and TsCl (1.79 g, 9.36 mmol, 1.5 eq), replacing the pyridine by tetramethylethylenediamine (TEMED) (1.4 mL, 9.36 mmol, 1.5 eq) as proposed by Yoshida [16]. The pure product is obtained as a colorless oil (2.06 g, yield = 93%). 1H-NMR (CDCl3, 400 MHz): 1.25 (t, J = 7.2 Hz, 3H); 1.27–1.40 (m, 2H); 1.52–1.71 (m, 4H); 2.25 (t, J = 7.4 Hz, 2H); 2.45 (s, 3H); 4.02 (t, J = 6.4 Hz, 2H); 4.08–4.16 (m, 2H); 7.35 (d, J = 8 Hz, 2H); 7.79 (d, J = 8.4 Hz, 2H).
tert-Butyl 4-(4-((6-ethoxy-6-oxohexyl)oxy)benzyl)piperazine-1-carboxylate 4 [
17]
tert-Butyl 4-(4-hydroxybenzyl)piperazine-1-carboxylate 2 (750 mg, 2.55 mmol, 1 eq), ethyl 6-(tosyloxy)hexanoate 3 (1.52 g, 5.10 mmol, 2 eq) and K2CO3 (885 mg, 6.40 mmol, 2.5 eq) are dissolved in 15 mL of acetone and placed under argon. The mixture is stirred in sealed tube 3 h at 130 °C in microwave oven. The solvent was removed under reduced pressure and the crude product was purified by column chromatography (eluent: CH2Cl2/AcOEt 3:2). The pure product is obtained as colorless oil (839 mg, yield = 78%). 1H-NMR (CDCl3, 400 MHz): 1.26 (t, J = 7.2 Hz, 3H); 1.45 (s, 9H); 1.47–1.55 (m, 2H); 1.64–1.83 (m, 4H); 2.27–2.40 (m, 6H); 3.41 (t, J = 5 Hz, 4H); 3.44 (s, 2H); 3.94 (t, J = 6.4 Hz, 2H); 4.06–4.16 (m, 2H); 6.83 (d, J = 8.8 Hz, 2H); 7.19 (d, J = 8.8 Hz, 2H).
Ethyl 6-(4-(piperazin-1-ylmethyl)phenoxy)hexanoate hydrochloride 5
tert-Butyl 4-(4-((6-ethoxy-6-oxohexyl)oxy)benzyl)piperazine-1-carboxylate 4 (182 mg, 0.43 mmol, 1 eq) is stirred in 2 mL of HCl (1 M in Et2O, 4.7 eq) for 30 min. The solvent is evaporated and dried under vacuum. The pure product is obtained as a white solid (154 mg, yield = 97%). 1H-NMR (MeOH-d4, 400 MHz): 1.21 (t, J = 7.2 Hz, 3H); 1.37 (t, J = 7 Hz, 2H); 1.43–1.53 (m, 2H); 1.59–1.70 (m, 2H); 1.73–1.82 (m, 2H); 2.32 (t, J = 7.4 Hz, 2H); 3.34–3.70 (m, 8H); 3.99 (t, J = 6.4 Hz, 2H); 4.04–4.12 (m, 2H); 4.33 (s, 2H); 7.00 (d, J = 8.8 Hz, 2H); 7.45 (d, J = 8.8 Hz, 2H).
6-(4-(Piperazin-1-ylmethyl)phenoxy)hexanoic acid hydrochloride 6:
tert-Butyl 4-(4-((6-ethoxy-6-oxohexyl)oxy)benzyl)piperazine-1-carboxylate 4 (2.27 g, 5.39 mmol, 1 eq) is stirred in 22 mL of HCl (1 M in Et2O, 4 eq) for 1 night, a solid appears. The mixture is evaporated then redissolved in 22 mL of HCl in Et2O (1M). The mixture is sonicated and stirred for 4h. As the reactant was still present, the mixture was extracted with CH2Cl2 and H2O. The organic layer gave the reactant after evaporation, and the aqueous layer gave the product. The pure product is obtained as a white solid (646 mg, yield = 36%). 1H-NMR (MeOH-d4, 400 MHz): 1.45 (s, 9H); 2.39 (t, J = 5 Hz, 4H); 3.43 (t, J = 5.2 Hz, 4H); 3.45 (s, 2H); 6.75 (d, J = 8.4 Hz, 2H); 7.25 (d, J = 8.4 Hz, 2H) HRMS (ESI+): Calcd for [C17H26N2O3]: 306.1943, found: 307.2012, correspond to (5b) + H+.
2-((E)−2-((E)−2-Chloro-3-(2-((E)−1,3,3-trimethylindolin-2-ylidene)ethylidene)cyclohex-1-en-1-yl)vinyl)−1,3,3-trimethyl-3H-indol-1-ium iodide 7 [
18,
19,
20]
2-Chlorocyclohex-1-ene-1,3-dicarbaldehyde (100 mg, 0.58 mmol, 1 eq), 1,2,3,3-tetramethyl-3H-indol-1-ium iodide (385 mg, 1.24 mmol, 2.13 eq) and sodium acetate (48 mg, 0.58 mmol, 1 eq) are dissolved in 3 mL of EtOH and put under argon. The mixture is stirred in a sealed tube 20 min at 120 °C in a microwave oven. The obtained solid is filtrated and washed with Et2O, then adsorbed on silica and purified by column chromatography (eluent: CH2Cl2/MeOH 9:1). The pure product is obtained as a green solid (300 mg, yield = 85%).1H-NMR (CDCl3, 400 MHz): 1.72 (s, 12H); 1.98 (qu, J = 6.0 Hz, 2H); 2.75 (t, J = 6.4 Hz, 4H); 3.72 (s, 6H); 6.20 (d, J = 14.4 Hz, 2H); 7.15 (d, J = 8.0 Hz, 2H); 7.20–7.27 (m, 2H); 7.34–7.42 (m, 4H); 8.34 (d, J = 14.4 Hz, 2H) HRMS (ESI+): Calcd for [C32H36ClN2]: 483.2562, found: 483.2569.
2-((E)−2-((E)−2-(4-(4-((6-Ethoxy-6-oxohexyl)oxy)benzyl)piperazin-1-yl)−3-(2-((E)−1,3,3-trimethylindolin-2-ylidene)ethylidene)cyclohex-1-en-1-yl)vinyl)−1,3,3-trimethyl-3H-indol-1-ium iodide 8 [
9]
Compound 4 (225 mg, 0.63 mmol, 1.5 eq) and TEMED (0.12 mL, 8.84 mmol, 2 eq) are dissolved in 2 mL of dimethylformamide (DMF) and placed under argon. The mixture is stirred at RT for 10 min. Compound 7 (257 mg, 0.42 mmol, 1 eq) is added and the mixture is stirred in a sealed tube for 1 h at 150 °C in microwave oven. After evaporation of the solvent under reduce pressure, the crude product is adsorbed on silica and purified by column chromatography (eluent: from CH2Cl2 to CH2Cl2/MeOH 9:1). The pure product is obtained as a blue solid (100 mg, yield = 26%).1H-NMR (CDCl3, 400 MHz): 1.24 (t, J = 6.8 Hz, 3H); 1.41 (s, 4H); 1.61 (s, 12H); 1.63–1.87 (m, 10H); 2.32 (t, J = 7.2 Hz, 2H); 2.49 (t, J = 6.4 Hz, 4H); 2.61–3.01 (m, 6H); 3.52 (s, 5H); 3.60–3.87 (m, 8H); 3.97 (t, J = 6.4 Hz, 2H); 4.11 (q, J = 7.2 Hz, 2H); 5.78 (d, J = 9.2 Hz, 2H); 6.90 (d, J = 8.4 Hz, 2H); 7.00 (d, J = 8.0 Hz, 2H); 7.12 (t, J = 7.6 Hz, 2H); 7.26–7.38 (m, 6H); 7.59 (d, J = 13.2 Hz, 2H) 13C-NMR (CDCl3, 100 MHz): 0.08, 14.34, 21.83, 24.79, 25.22, 25.76, 26.99, 28.99, 34.31, 47.98, 55.19, 60.36, 67.78, 96.36, 109.33, 114.50, 121.97, 123.57, 124.53, 128.58, 130.84, 130.96, 140.08, 141.28, 143.41, 173.74 FTMS (ESI+): Calcd for [C51H65N4O3]+: 781.5051, found: 781.5046.
2-((E)−2-((E)−2-(4-(4-((5-Carboxypentyl)oxy)benzyl)piperazin-1-yl)−3-(2-((E)−1,3,3-trimethylindolin-2-ylidene)ethylidene)cyclohex-1-en-1-yl)vinyl)−1,3,3-trimethyl-3H-indol-1-ium iodide CyB
6-(4-(Piperazin-1-ylmethyl)phenoxy)hexanoic acid hydrochloride (213 mg, 0.65 mmol, 4 eq) and TEMED (0.12 mL, 0.80 mmol, 5 eq) are added in 10 mL of DMF. The mixture is stirred for 10 min. 2-((E)−2-((E)−2-chloro-3-(2-((E)−1,3,3-trimethylindolin-2-ylidene)ethylidene)cyclohex-1-en-1-yl)vinyl)−1,3,3-trimethyl-3H-indol-1-ium iodide (7) (100 mg, 0.16 mmol, 1 eq) is then added to the mixture and stirred at RT for 2 h 30. The mixture is extracted with AcOEt and the organic layer is washed 6 times with brine. The aqueous layer is reextracted once with AcOEt. As the limiting reactant was still present, the organic layer was then evaporated. The obtained residue was dissolved again in 5 mL of DMF. TEMED (0.12 mL, 0.80 mmol, 5 eq) and 6-(4-(piperazin-1-ylmethyl)phenoxy)hexanoic acid hydrochloride (213 mg, 0.65 mmol, 4 eq) were added to the mixture. The mixture was stirred 3 days at RT. The mixture was then extracted with AcOEt and the organic layer washed with brine. The organic layer was evaporated and dissolved in a minimum of CH2Cl2 and placed in centrifugation tubes. Then Et2O was added, and the mixture was centrifugated. The liquid was removed and the centrifugation process repeated several times, until the liquid stays limpid. The product (CyB) was obtained as a blue solid (134 mg, yield = 92%). 1H-NMR (CDCl3, 400 MHz): 1.50–1.74 (m, 6H); 1.59 (s, 12H); 1.81 (qu, J = 6.4 Hz, 4H); 2.47 (t, J = 6.4 Hz, 4H); 2.81 (s, 4H); 3.51 (s, 6H); 3.71–3.83 (m, 6H); 3.97 (t, J = 6.4 Hz, 2H); 5.77 (d, J = 13.6 Hz, 2H); 6.91 (d, J = 8.4 Hz, 2H); 7.03 (d, J = 7.6 Hz, 2H); 7.14 (t, J = 7.6 Hz, 2H); 7.24–7.36 (m, 6H); 7.59 (d, J = 13.4 Hz, 2H) 13C-NMR (CDCl3, 100 MHz): 21.82, 24.77, 25.15, 25.72, 28.93, 29.08, 29.78, 31.41, 34.44, 48.02, 53.55, 53.70, 54.73, 62.01, 67.83, 96.45, 109.43, 114.40, 114.60, 122.00, 123.72, 124.56, 127.20, 128.61, 130.69, 131.24, 140.10, 141.42, 143.33, 158.86, 169.58, 173.33 HRMS (ESI+): Calcd for [C49H61N4O3]+: 753.4738, found: 753.4732.
2.3 Preparation of polymer films
Polymer films have been made using a mixture of CH2Cl2, HydroMed-D4™ (10 wt%) and fluorophore placed in a Teflon mold and let to dry under a CH2Cl2 atmosphere to limit the formation of bubbles inside the film. The same solution is used to make several films each time, as the mold has 6 holes to prepare the films (0.15 mm thick). The film has been then placed in water to inflate (up to 0,18 mm thick) and cut with a circular punch to give a disc which has been used for spectroscopy measurements. The disc is then placed in a home-designed holder to keep it vertically perpendicular to the light beam inside the quartz cuvette (see Figure S2 in supplementary information). Each measurement has been made 5 min after the change of the solution in the quartz cuvette to let the water diffuse inside the film. The absorbance measurements required the use of an integrating sphere as the diffusion gave non-exploitable spectra without it.
A solution of 1 mg of CyB in 100 mL of CH2Cl2 + 14.8 g of HydroMed D4 (to have a solution of 10% wt in HydroMed D4) was prepared but gave too concentrated films (reaching the detection limit in absorbance). So this solution has been diluted 10 times with CH2Cl2 + HydroMed D4 (10% wt), to only dilute CyB in the resulting films.
2.4 Calculation
Geometry optimizations of CyB and CyBH+ were done at the b3lyp/6-311G + (d,p) level of calculation followed by a frequency job to ascertain that a true minimum was obtained. Theoretical UV–Visible spectra were calculated on optimized geometries structures by an energy calculation using time dependant DFT calculation at TD PBE0/6–311 + g(d,p) level and solving on 18 first singlet states. A standard solvation model (IEFPCM) for water was used. The molecular orbitals were subsequently obtained at the same level. The calculations were done with the Gaussian 16 (Revision B.01) software [21] and the molecular orbitals visualized with GaussView 6.1.1 (isovalue 0.02). Summary of geometry, cartesian coordinates of optimized geometry and normal modes for both CyB and CyBH + are given in Supplementary Information Section.
2.5 Spectroscopic measurements
Absorbance spectra were measured with Agilent Technologies Cary 100 UV–Vis and a Shimadzu UV-2600 UV–Vis spectrophotometer equipped with an ISR-2600Plus integrating sphere (for film absorbance measurements). Fluorescence spectra were measured with a Horiba Jobin Yvon Fluoromax®−4 spectrofluorometer and a Shimadzu RF-6000 spectrofluorophotometer. The quartz cuvettes were purchased from Helma or Thuet. Slits were adjusted depending on the sample (2–5 nm). Scientific data analysis and spectral processing were conducted on Origin 2018 graphing and analysis software—b.9.5.0.193 (academic) by OriginLab Corporation (www.originlab.com).
All the spectra were recorded at room temperature using 10-mm path-length quartz cells, either in EtOH or dimethyl sulfoxide (DMSO), or in aqueous mixture of pH buffer and EtOH or DMSO (9:1 or 8:2, v:v) to record the spectra following the pH. The concentrations were adjusted to be at 0.1 in absorbance at the maximum for recording the emission spectra, to limit inner filter effect. For the aqueous solutions, the concentration has been adjusted so that the maximum of absorbance is 0.1 for the acidic conditions, and kept the same with basic conditions to have comparable data. pH buffers were prepared by dissolving citric acid and sodium phosphate in MilliQ water to get buffers at pH 2.6, 3.0, 3.6, 4.0, 4.6, 5.0, 5.6, 6.0, 6.6, 7.0, 7.6 and 8.0 (see Table S1 in supplementary information for the details). Molar extinction coefficients have been calculated with different dilutions from a same mother solution. Fluorescence Quantum Yields (ΦF) between 700 and 850 nm were calculated with HITC in EtOH (ΦF = 0.28) as reference [22]. pKa were calculated thanks to the method described by Nguyen et al. [23] using solutions of pH buffer and EtOH or DMSO (9:1 or 8:2, v:v).
2.6 Bacteria toxicity tests
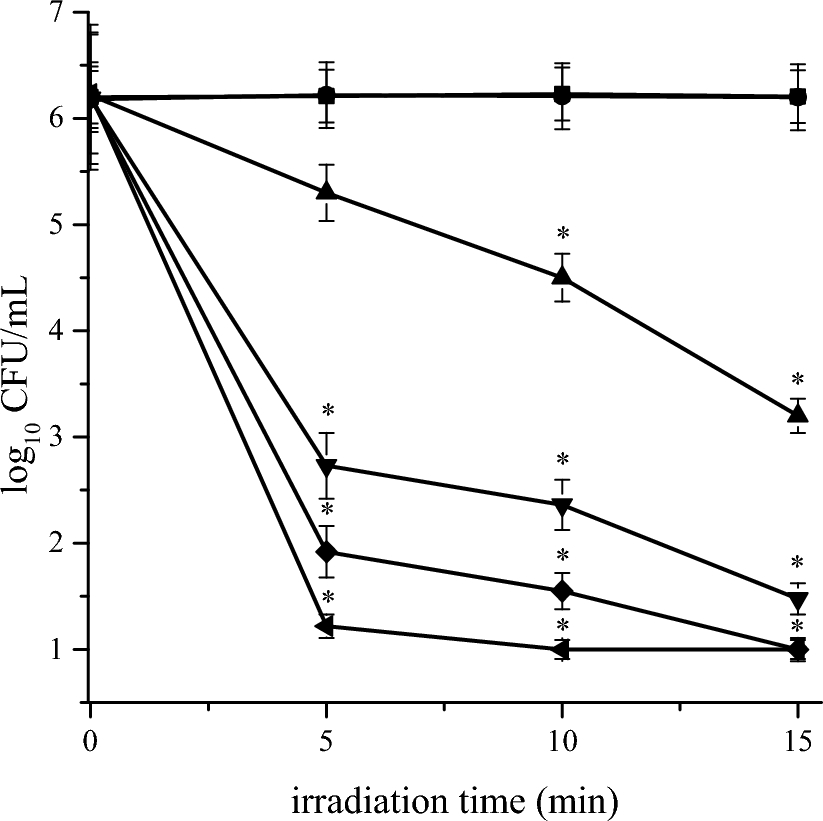
The bacteria used for the toxicity test come from the same strain E. coli BW2511.3 kept at − 80 °C. These bacteria are then dispersed over LB-Agar on a round petri dish and grown at 37 °C for 16 h. Then one colony is taken and put to grow in 10 mL of LB solution (MilliQ water with Luria–Bertani broth (LB) powder, 2,5% w) in an erlenmeyer at 37 °C with agitation at 120 rpm overnight, to keep a homogeneous solution. 100 µL of this solution were taken to make a solution diluted 100 times (considering bacteria) in another erlenmeyer with LB solution to make the growth experiment sample. The bacterial concentration is then followed over time by measuring the absorption of the solution at 600 nm.
Toxicity of CyB has been investigated in solutions with E. coli BW2511.3: 300 µg of CyB (3.41 × 10–7 mol) were dissolved in 1.6 mL of DMSO, and then diluted in 14.4 mL of LB solution (final concentration of CyB: 2.13 × 10–5 M). Then E. coli BW2511.3 were added to the solution (dilution 100 times regarding bacteria). The control experiment has been conducted in the same conditions, but without CyB. The absorbance of the bacteria has been measured at 600 nm every 25 min over 6 h. For the bacteria in solution with CyB, each measure has been conducted as follow: 1 mL of the solution was taken and placed in an Eppendorf tube, then a centrifugation (2500 rpm, 10 min) has been conducted to remove the colored liquid, then the bacteria has been redispersed in 1 mL of MilliQ water with LB and the absorption at 600 nm has been measured. This protocol allows us to prevent an overestimation of the absorption due to the absorbance of CyB at 600 nm.
2.7 Practical application on yogurt
Yogurts are made from a mixture of commercial yogurt (Danone) and milk in proportions of 1:8. The mixture is then left for 14 h at a temperature of 37 °C leading to the formation of yogurt with a pH of 4.75. The pH of the milk is 6.80. The milk or the yoghurt is then placed in a crystallizer with a wedge to hold the hydromed D4/CyB sample in the same position. The fluorescence of the film is measured using a fiber optic bundle [24].



Comments (0)