
Remember me






Inflammation is a fundamental element of our immune response to any kind of stress, such as injury, infection, etc. A growing number of studies have broadly examined the link between inflammation and cancer, supporting its role in promoting tumor initiation, progression, and metastasis. While acute inflammation has a protective role in eliminating harmful stimuli and initiating tissue repair, chronic inflammation creates a pro-tumorigenic microenvironment characterized by the sustained production of inflammatory mediators, including reactive oxygen species (ROS), chemokines, and cytokines. These molecules enhance the invasiveness and metastatic potential of cancer cells, stimulate cell proliferation, promote angiogenesis, and block apoptosis [10].
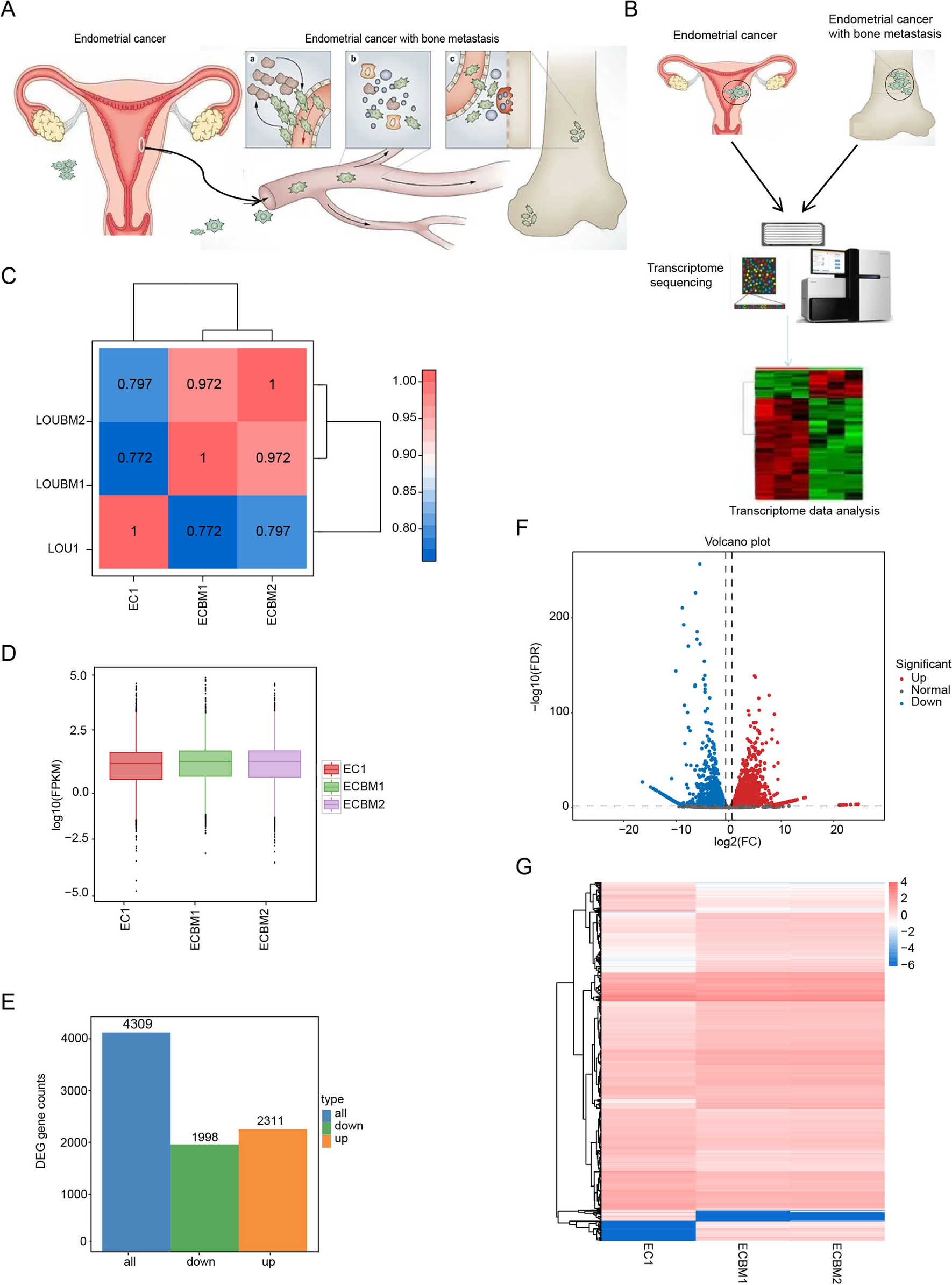
Chronic bacterial infections are a major factor that leads to chronic prostatitis, creating an inflammatory environment in the prostate, which can trigger cancer development [11]. Several intrinsic and environmental factors, such as bacterial infections, viruses, diet, age, hormones, autoimmune reactions, environmental exposure, and certain medications, increase the risk of chronic prostate inflammation (Fig. 1). Prolong inflammation caused by these factors can drive molecular changes that activate oncogenes or deactivate tumor suppressor genes, leading to the development and progression of PC.
Fig. 1
Factors contributing prostate inflammation: Various intrinsic and environmental factors contribute to the risk of chronic prostatic inflammation, including bacterial infections, viral infection, dietary factors, aging, hormonal imbalance, autoimmune responses, environmental exposers or certain medications
3.1 Factors contributing to chronic inflammation and the development of prostate cancerChronic bacterial infections are a major factor that leads to chronic prostatitis, creating an inflammatory environment in the prostate, which can trigger cancer development [11]. Several factors, such as bacterial infections, viruses, diet, age, hormones, autoimmune reactions, environmental exposure, and certain medications, increase the risk of chronic prostate inflammation (as seen in Fig. 1). These factors persist over time, causing genetic and epigenetic changes. These changes activate oncogenes (cancer-causing genes) and deactivate tumor suppressor genes, which drive PC development and progression. In addition, chronic inflammation can promote tumor heterogeneity through enhancing genetic divergence of tumor cells, such as TMPRSS2: ERG fusion. Inflammation has been proposed as a possible mechanism of this fusion by generating oxidative stress, which leads to damage in the form of double-strand breaks in DNA, ultimately enabling the occurrence of gene fusions [12]. Further inflammatory TME also serves as a selective pressure for tumor cell variants through genomic instability and heterogeneity [13]. As a result, there can be different subtypes of tumors that differ in aggressiveness, response to therapy, and tendency to metastasize.
3.1.1 Pathogenic infectionsBacterial infection contributes directly to chronic inflammation in the prostate, increasing the chances of PC. Common bacteria, including E. coli (55.5% of cases), Enterobacter cloacae, Klebsiella pneumoniae, Pseudomonas, and Aeruginosa, infect the prostate by entering through the urethra, infected urine backflow, or spreading from the rectum. These chronic bacterial infections can cause prolonged inflammatory cytokine production by chronic activation of NF- κβ, leading to tumorigenesis [14]. In a study by Sfanos in 2008, found that most prostatectomy samples (87%) contain bacterial DNA from one or more species. However, the majority of individual tissue core samples are negative, suggesting regional heterogeneity in the presence of bacteria. The presence of bacteria and resultant inflammation may create a diverse TME that drives the evolution of tumor subtypes [15].
Viruses also play a role in causing chronic prostate inflammation and increasing PC risk. HPV (Human Papillomavirus), EBV (Epstein-Barr Virus), HSV (Herpes Simplex Virus), XMRV (Xenotropic Murine Leukemia Virus-Related Virus), and polyomaviruses (BKV, JCV, and SV40) are among the viruses detected in prostate tissue with known cancer-causing potential [16].
3.1.2 Hormonal imbalancesAndrogens, such as testosterone and dihydrotestosterone (DHT), are known for playing a key role in normal growth and functioning of the prostate gland. These androgens are bound to androgen receptors (AR) in prostate cells, which regulates the cell growth and survival. And change in androgen levels is associated with initiation and progression of PC [17]. In PC, tumor growth is sustained through AR signaling activated by androgens. As a result, androgen deprivation therapy (ADT) is clinically used for PC treatment, with its mechanism involving the lowering of androgen levels or the blocking of AR activity. However, PC cells tend to preserve AR signaling even when androgen levels drop. They achieve this by upregulating AR expression, or producing AR variants that function without androgens [18]. These adaptations allow cancer to keep growing despite ADT, leading to castration-resistant PC (CRPC). Understanding these adaptive mechanisms is crucial for the development of more effective therapeutic strategies that can target the unique characteristics of each tumor.
3.1.3 Dietary habitsConsuming diets that are high in saturated fats, processed meats, have been reported significantly for increasing systemic inflammation, while consuming foods rich in omega-3 fatty acids have been seen to lower the risk of PC [19]. Obesity is associated with chronic low-grade inflammation and hormonal changes and this can increase risk for PC development [20]. In a study of 525 men recently diagnosed with PC, researchers found a connection between drinking high-fat milk and the progression of the cancer [21]. Moreover, a observational study found that intense exercise was linked to a decreased risk of advanced PC, as well as it decreases the chance of TMPRSS2 fusion-positive cases [22]. A gene set enrichment analysis of prostate tissue adjacent to tumors from same study indicated that immune pathways in the TME were altered in men who engaged in vigorous exercise compared to those who did not [23]. Recent research has shown that consuming a lot of macronutrients might raise oxidative stress and hence cause inflammation. It is important to talk about dietary carbohydrates because they can cause long-term effects from nutritionally driven oxidative stress [24]. Researchers have focused extensively on carbohydrate consumption because of its link to PC with high glycemic load or glycemic index diets [25]. A meta-analysis and systematic review and of 59 studies with 280,199 patients revealed that obesity raises the risk of PC-specific death by 19% and overall mortality by 9% [26]. Hyperinsulinemia is linked to various cancers, including PC. Studies, both in vivo and in vitro, have shown that high grade prostate tumors exhibit elevated levels of the insulin receptor-A isoform and a greater number of insulin receptors [27, 28]. These studies highlight the importance of diet, exercise, and metabolic health in preventing risk of PC.
3.1.4 AgingAging reduces tissue mass and decreases the functionality of adult stem cells in many tissues. However, in the case of prostate, the gland grows during two main periods: the first during puberty and the second in a man’s thirties. With the increase in age in men, the prostate typically enlarges, often reaching the size of a lemon by the age of 60. The exact cause for the prostate enlargement is unclear. Some researchers link this growth to an increase in luminal progenitor cells in the prostate.
3.1.5 Environmental exposuresExposure to environmental toxins, pollutants, and carcinogens has been linked to prostatic inflammation and an increased risk of PC. Mainly, tobacco smoke and pesticides, contribute to this risk by generating excessive oxidative stress, leading to DNA damage, and PC development and malignancy [29]. Moreover, exposure to bisphenol A, benzo(a)pyrene, and ethyl-paraben have been associated with increased risk of PC [30].
3.1.6 Immunosuppressive medicationsImmunosuppressive medications, including anti-inflammatory drugs (AIMs) like non-steroidal anti-inflammatory drugs (NSAIDs), immunosuppressants, and glucocorticoids, play a role in the development of PC, particularly in cases of metastatic PC. Some studies link NSAIDs, especially aspirin, to a lower risk of PC, while others suggest that using AIMs—particularly non-aspirin NSAIDs, Coxibs, and acetaminophen—may increase PC risk [31].
Therefore, identifying key regulators and pathways of inflammation is essential to understanding its role in PC and advancing treatment strategies.
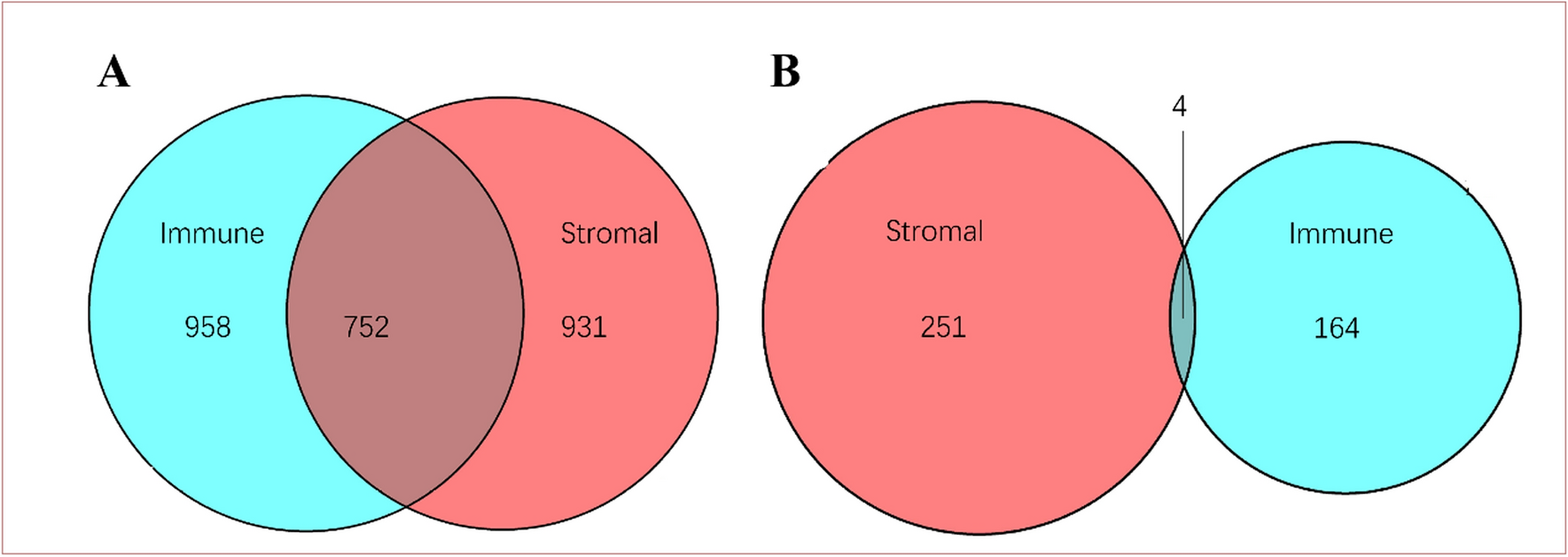
3.2 Molecular pathways involved in inflammation-induced prostate cancerThe development of cancer via chronic inflammation involves several pathways, including NF-κB, PI3K/AKT, and JAK/STAT. These pathways regulate immune responses, cell proliferation, survival, and metastasis. When these pathways become dysregulated, they can contribute to cancer progression (Fig. 2).
Fig. 2
Inflammatory pathways implicated in prostate cancer: NF-κB, activated by inflammatory triggers like TNF-α, ROS among others, plays a central role in the release of cytokines, leading to chronic prostate inflammation that can lead to PC development. Similarly, the PI3K/AKT pathway, when activated, promotes the secretion of pro-inflammatory cytokines, contributing to the inflammation mediated PC. The JAK/STAT pathway, stimulated by cytokines, interferons, further drives inflammation, metastasis, and lineage plasticity, all of which are pivotal in the progression of prostate cancer
3.2.1 NF- κβ: a central player in inflammation and cancerBoth innate and adaptive immunity depend greatlyon NF-κβ, a major regulator of inflammatory reactions. NF-κβ controls the production of chemokines and cytokines, anti-apoptotic proteins, and adhesion molecules in immune cells and it is the key activator of many pro-inflammatory genes. Inflammatory triggers that activate this pathway include both infectious and non-infectious stimuli. Toll-like receptors (TLRs), a protein present on the immune cell surface, play a crucial role in this process, by recognizing the pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), this recognition initiates a signaling cascade that activates NF-κβ. Non-infectious stimuli including ROS, cytokines like TNFα and IL-1β, and other stress signals can also cause NF-κβ activation [32, 33].
When NF-κβ stimulates innate immune system cells, such neutrophils and macrophages, which aid in the removal of infections and the start of tissue healing, pro-inflammatory mediators are produced. PAMPs, proinflammatory cytokines generated by these innate immunity cells, and certain TNFSF (tumor necrosis factor receptor superfamily) members bind to specific receptors in the TLR/IL-1R superfamily or the TNFRSF. The IKK complex (Iκβ kinase), which is made up of the three subunits IKKα, IKKβ, and IKKγ (NEMO-NF-κβ essential modulator), is activated by this binding, leading to NF-κβ activation, which drives the innate immune response [34].
NF-κB is also essential for adaptive immune responses; it controls the activation and differentiation of T and B cells. It also plays a role in the development of effector subsets, Th1, Th2, Th17, and regulatory T cells (Tregs), which is supported by co-stimulatory signals from molecules such as CD28. B cells multiply, develop into plasma cells, and release antibodies as a result of NF-κβ activation triggered by antigen detection by the B cell receptor (BCR) [35]. Furthermore, T cell co-stimulatory signals, which are mediated by cytokines like IL-4 and molecules like CD40 ligand, further boost B cell NF-κβ activation, encouraging antibody class flipping and differentiation.
However, persistent or dysregulated activation of NF-κβ can contribute to chronic inflammatory diseases, autoimmune diseases and cancer. In these situations, the negative feedback mechanisms that typically end the inflammatory response may be overpowered by the prolonged NF-κβ activation stimulus [36]. It is this complex signaling cascade that highlights the role of NF-κβ in controlling immunological responses, inflammatory reactions, and the development of cancer (Table 1).
Table 1 Therapeutic targeting of inflammation in Prostate cancerThe promotion of inflammation is mostly dependent on NF-κβ signaling, and chronic inflammation is a major risk factor for PC. Most cancer cells have aberrantly active NF-κβ, which is thought to be responsible for apoptosis suppression, angiogenesis promotion, epithelial-mesenchymal transition/metastasis facilitation, cellular metabolic alteration, anti-tumor immune response suppression, and accelerated cell proliferation.
The abnormal activation of NF-κβ in cancer cells is also linked to the development of malignant tumors in a number of cancer types, including HNSCC (Head and Neck Squamous Cell Carcinoma), breast cancer, colon cancer, and lung cancer [37,38,39,40]. Chronic inflammation is a key activator in the metastatic cascade.
Additionally, NF-κβ influences the cells recruited to and forming the tumor microenvironment. NF-κβ becomes activated in particular microenvironment cells upon exposure to PAMPs, endogenous TLR ligands, and unknown ligands through TLR and other receptors. For example, TLR signaling via MyD88-dependent pathways requires TNF receptor-associated factor 6 (TRAF6). When NF-κβ is activated in these cells, proinflammatory cytokines, such as TNF and IL-1, are produced [41]. These cytokines then trigger NF-κβ activation in premalignant or tumor cells, which promotes the synthesis of genes related to angiogenesis, metastasis, proliferation, and survival.
Moreover, EMT, a complex process involving the breakdown of cell–cell adhesion and the gain of mesenchymal traits by epithelial cells, is associated with increased cancer invasion and metastasis. And NF-κβ signaling has been identified as a key player in promoting EMT, in various cancers including PC, contributing to enhanced invasiveness and metastasis.
3.2.2 PI3K/AKT pathwayThis pathway is important for many cellular processes such as growth, proliferation, survival, motility, metabolism, and immune response regulation. Additionally, inflammation promotes the growth of tumor by stimulating phosphatidylinositol 3-kinase (PI3K) /protein kinase B (PI3K). pathway. For instance, PI3K/Akt signaling triggered by inflammation controls migration and permeability of endothelial cells and affects the course of tumor. By controlling the activity of downstream targets, the PI3K signaling pathway influences the secretion of inflammatory cytokines by cells of the innate immune system.
The primary signaling molecules that activate the PI3K pathway include oncogenes like Ras that attach to the p110 subunit of PI3K, G-protein-coupled receptors (GPCRs), and receptor tyrosine kinases (RTKs). When PI3K is engaged, its catalytic component changes PIP2 into PIP3. When PIP3 is produced, PI3K binds to the pleckstrin homology domain of Akt, resulting in conformational changes that phosphorylate Akt. This phosphorylation process is necessary for the full activation of Akt. Activated Akt moves to the cell membrane from the cytoplasm. NF-κβ and mTOR are two downstream molecular proteins that are either directly or indirectly activated by this cascade, increasing inflammatory responses, cell survival, proliferation, and PC malignancy [42]. The PI3K/AKT pathway plays a role in the development of Multi-Drug Resistance (MDR), partly because of activation of NF-κβ. This pathway may be involved in MDR by inducing PI3K/AKT/NF-κβ activation, which results in cyclin D1 transformation, G1/S phase protein expression, and cell cycle acceleration [42]. Furthermore, uncontrolled PI3K signaling is common in cancer, mainly due to the roles of its catalytic subunits, p110α and p110β. PC is one of several malignancies linked to mutations in the PI3KA gene, which codes for p110α. This gene is essential for endothelial cells because, in addition to controlling proliferation and the cell cycle, it encourages the development of blood vessels that are necessary for tumor growth and metastasis. Ablating p110β significantly reduces Akt activation, leading to a marked decrease in tumor growth in PTEN-deficient prostate cancer models. According to recent research, p110β disruption slows down the onset and development of CRPC, however, p110α inhibition has no effect on PTEN-null CRPC [43].
PTEN is a key regulator of the PI3K/AKT pathway, acting as a tumor suppressor by dephosphorylating PIP3 back to PIP2, which inhibits the pathway. PTEN’s phosphatase activity, which targets both lipids and proteins, helps prevent uncontrolled cell growth and promotes cell death [44]. PTEN mutations disrupt its tumor-suppressive role by either inactivating its phosphatase function or altering its protein-specific activity. Besides regulating PI3K/AKT, PTEN supports genomic stability, cell renewal, senescence, migration, and metastasis within the tumor microenvironment. Glioblastoma, lymphoma, breast, prostate, endometrial, ovarian, colon, and melanoma are among the cancers that have been linked to PTEN mutations [45]. Akt activation phosphorylates a wide range of substrates involved in angiogenesis, metabolism, and cell survival, such as TSC1, TSC2, GSK3, FOXO, p21, p27, caspase-9, BAD, and iNOS. PC and other cancers are characterized by hyperactivation of Akt. PC is caused by hyperactivation of the PI3K/AKT/mTOR system, which is brought on by overexpression of AKT or loss of PTEN in prostate cells, according to research using transgenic and mutant animal models. AKT1, TSC1, and TSC2 (Tuberous Sclerosis Complex 1 and 2), which encode the proteins hamartin and tuberin, respectively, are crucial in PTEN-mediated cancer progression [46]. In particular, it has been demonstrated that PTEN deletion, in conjunction with loss of mTOR or RICTOR (a component of the mTORC2 complex), slows the course of PC [47]. Up to 42% of primary and all metastatic PC samples have aberrant gene expression and changes in PI3K pathway components, based on genomic and transcriptome analyses [48]. PTEN loss and subsequent pathway activation are observed in 40% of primary tumors and 70% of metastatic cases, highlighting the significant role of the PI3K/AKT/mTOR pathway in PC.
3.2.3 JAK/STAT signaling pathwayIn PC, abnormal and prolonged Janus kinase/signal transducer and activator of transcription (JAK/STAT) activation is associated with tumor development, progression and lineage plasticity. The JAK/STAT signaling system may be activated by a variety of cytokines, growth factors, and hormones, including growth hormone, interleukins, and interferons. This can result in phenotypic changes in a range of tissues and cell types. JAK is transphosphorylated when a ligand attaches to its receptor. Tyrosine residues on the receptor are subsequently phosphorylated by the active JAK, forming a docking site for STAT proteins. Then, through SH2-domain–phospho-tyrosine interactions, JAK phosphorylates STATs at this location, causing them to separate from the receptor and form homodimers or heterodimers. After that, these dimers go to the nucleus, where they attach to the promoters of target genes and control transcription.
Among other ligands, IL-6 stands out as a potent inflammatory cytokine and a crucial regulator of PC progression via JAK/STAT pathway. Elevated serum IL-6 levels have been observed in metastatic PC and CRPC cases compared to healthy men or those with localized disease [49]. A recent study found that, in mice model, a high fat diet elevated IL-6 secretion by prostate macrophages, which triggered STAT3-driven growth of myeloid-derived suppressor cells and fostered a tumor-promoting [50]. Apart from IL-6, there are other drivers that involve the same pathway for induction of PC via inflammation, which includes IL-11, IL-8, Oncostatin M (OSM), and Leukemia inhibitory factor (LIF). OSM and LIF are primarily found in the stromal compartment of the prostate; however, their expression is found to be elevated in the prostate epithelial cells in PC patients [51]. OSM is responsible for EMT, induction of morphological changes, and migration of PC cells through the JAK/STAT3 pathway [52], while ADT- induced LIF, activates STAT3 signaling to promote neuroendocrine differentiation (NED) and CRPC [53]. Similarly, IL-8 plays role in preventing apoptosis in PC cells through the STAT3 pathway [54]. And IL-11 activates STAT3 by binding to its receptor (IL-11Rα), which promotes the proliferation of PC cells leading to oncogenesis [55]. Additionally, studies indicate that JAK/STAT signaling is vital for driving lineage plasticity, a factor that contributes to resistance against androgen receptor (AR)-targeted therapies in PC [56].
3.3 Oxidative stress and reactive oxygen species (ROS)ROS are chemicals produced by immune cells during a normal inflammatory response to kill pathogens. Elevated ROS levels in cells can damage DNA, increasing the risk of mutations that could lead to cancer. NADPH oxidases (Nox) are crucial for both generating ROS and supporting the growth and maintenance of malignant cells, and significant ROS production takes place in PC [57, 58]. ROS are continuously produced by the body due to immunological reactions, mitochondrial bioenergetics, and oxidative metabolism. Physiological activation of AR has been shown to promote the formation of ROS [59]. This implies that androgen activation in PC cells may contribute to mt-DNA mutations and aging-related processes by increasing ROS formation, which can damage DNA and perhaps influence the mutation rate in cells, including PC cells.
The glutathione peroxidase system eliminates peroxides through the glutathione redox system. This serves as a crucial antioxidant defense mechanism. And glutathione acts as a marker for redox status in various diseases, aging processes, and cell death. It utilizes glutathione peroxidase to catalyze the glutathione redox system to neutralize peroxide [60]. PC mainly inactivates glutathione peroxidase 3 (GPX3), an enzyme that is dependent on selenium and essential for the detoxification of reactive oxidative species. Homozygous and hemizygous deletions of the GPX3 gene were shown to be common in PC samples in 39% of the examined samples. Additionally, Yu et al. found that 90% of the GP X3 exon 1 region is methylated in PC samples [61]. The progression from inflammation to preneoplastic lesions, such as high-grade prostatic intraepithelial neoplasia (PIN), and eventually to prostate cancer (PC) may be influenced by the methylation of the GSTP1 gene. This methylation leads to a loss of the gene's protective function [62].
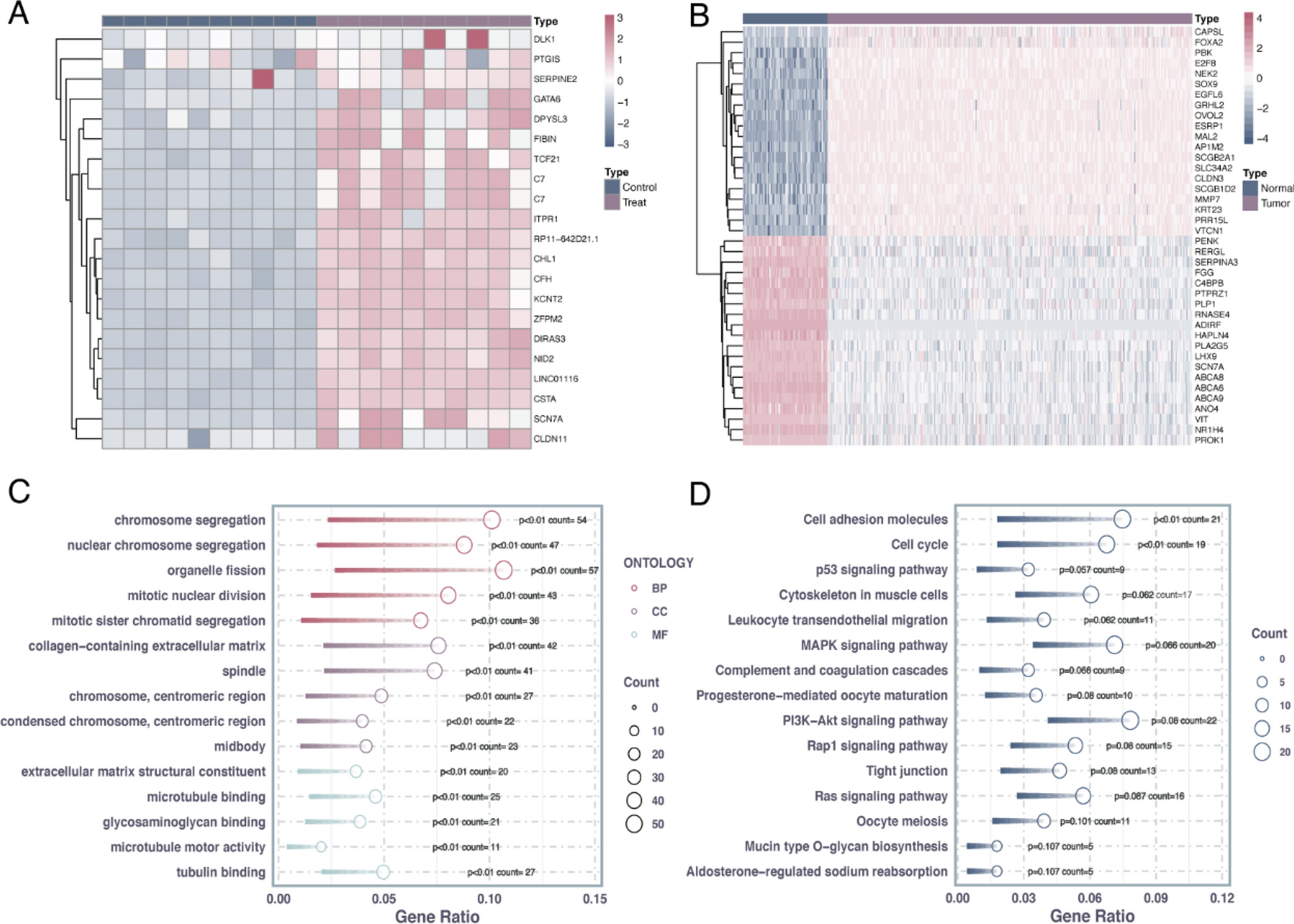
3.4 Pro-inflammatory tumor microenvironmentThe tumor microenvironment (TME) of PC is a complex ecosystem that includes tumor cells, stromal cells, immune cells, and a dense ECM. This intricate network of cellular and non-cellular components supports tumor development and progression while also contributing to drug resistance. The prostate TME is characterized by a pro-inflammatory environment that influences cancer cell behavior and surrounding stroma. Inflammation results from factors such as chemokines, cytokines, inflammasomes, stromal cells, and fibroblasts secreted by tumor cells and immune cells. This inflammatory setting promotes pro-tumorigenic processes like angiogenesis, invasion, and metastasis (Fig. 3).
Fig. 3
The pro-inflammatory tumor microenvironment of PC, a complex ecosystem of tumor cells, stromal cells, immune cells, cytokines, chemokines, pro-inflammatory mediators, inflammasomes, and a dense extracellular matrix, which collectively drive tumor progression, metastasis and immune evasion
3.4.1 Growth factorsGrowth factors stimulate signaling pathways that allow cancer cells to survive and adapt in the TME. Important growth factors in PC include IL-6, transforming growth factors (TGF-α and TGF-β), epidermal growth factor (EGF), and insulin-like growth factors I and II (IGF-I and IGF-II). Among these growth factors, the overexpression of epidermal growth factor receptor (EGFR) is particularly significant, as it is linked to lower survival rates in PC patients. As PC advances, cells shift from producing EGF to TGF-α, promoting autocrine growth and uncontrolled proliferation [63]. High levels of circulating IGF-I are associated with an increased risk of PC [64]. IL-6 also affects growth, differentiation, and apoptosis in PC by activating the STAT and MAPK signaling pathways. Patients with metastatic PC typically have higher serum levels of IL-6, which may speed up tumor growth over time.
3.4.2 Proinflammatory mediatorsPGE2 is the most abundant proinflammatory mediator in prostate tissue, with elevated levels in PC [65]. PGE2 is produced by the COX pathway from arachidonic acid and stimulates the growth, multiplication, and metastasis of cancerous cells in addition to upregulatin
Comments (0)