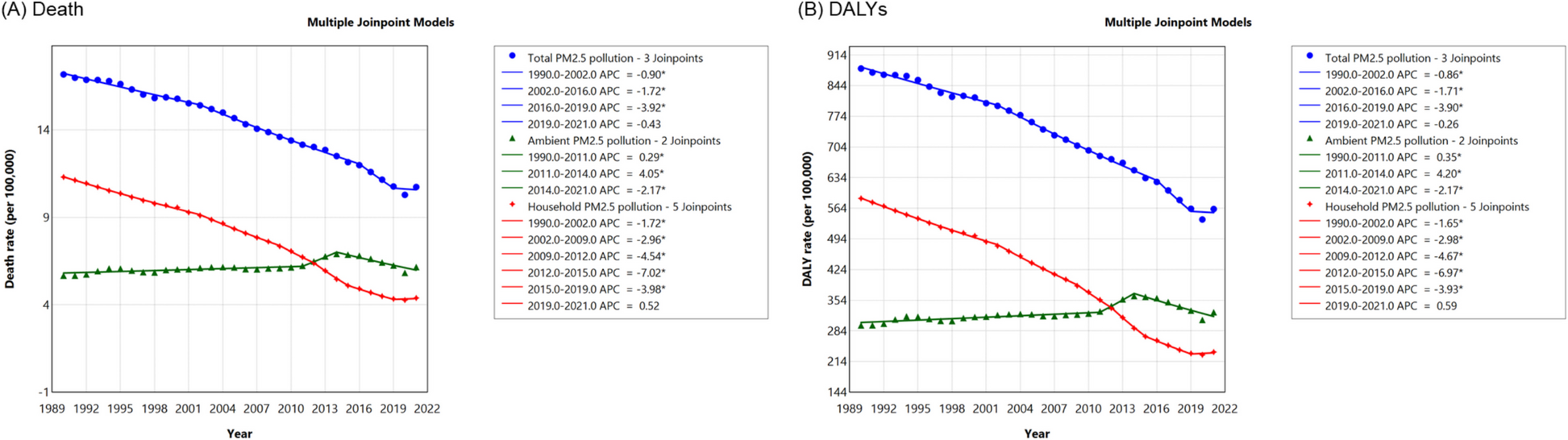
Using a large multicenter prospective cohort of 5244 AMI patients, we identified a “U-shaped” association between LDL-C levels and mortality risks after multivariable adjustment. However, this association was found to be mediated by hsCRP levels. The U-shaped pattern was observed exclusively in patients with hsCRP ≥ 3 mg/L, whereas high LDL-C levels were positively associated with increased mortality risks in patients with hsCRP < 3 mg/L.
Hypercholesterolemia is a well-established risk factor for the development and progression of atherosclerotic cardiovascular disease. Randomized controlled trials on statins and non-statin lipid-lowering agents, including ezetimibe, PCSK9 inhibitors, inclisiran, and bempedoic acid, have consistently shown the effect of cardiovascular event reduction [20]. Therefore, lipid-lowering therapy is recommended for almost all CAD patients. For patients with AMI, according to the 2023 ESC guidelines for the management of ACS, the LDL-C treatment goal for secondary prevention should aim to achieve levels less than 1.4 mmol/L, with lipid-lowering therapy advised to be initiated as early as possible [4]. However, the prognostic impact of baseline LDL-C levels on clinical outcomes in patients with cardiovascular diseases remains controversial, leading to an ongoing debate regarding the intensity of LDL-C control. A study based on a large-scale nationwide cohort suggested that the association between LDL-C levels and all-cause mortality was U-shaped in the general population [21]. Additionally, a recent collaborative study involving three randomized trials found a neutral association between baseline LDL-C and cardiovascular events among patients receiving statins [22]. In terms of AMI, the relationship between LDL-C and mortality appears paradoxical according to several large-scale cohort studies. Reddy et al. observed that low LDL-C levels were associated with a higher risk of in-hospital mortality among 115,492 AMI patients based on data from the US National Registry of Myocardial Infarction [8]. This phenomenon, known as the “lipid paradox,” still exists in terms of long-term outcomes. A study based on the SWEDEHEART registry reported an inverse association between LDL-C and long-term mortality in 63,168 patients hospitalized for a first AMI [7]. Similarly, another Korean study of 5532 AMI patients found that those with baseline LDL-C < 70 mg/dL had a significantly higher risk of long-term cardiovascular events, even after more than 3 years of statin therapy [23].
Several studies suggested that this phenomenon might be due to inadequate adjustment for confounders, such as comorbidities or in-hospital medical therapies. For example, a study of 9571 AMI patients undergoing PCI reported that the independent association between LDL-C levels and 1-year mortality risk diminished after adjusting for demographic and biological variables [24]. This study also adjusted for inflammatory biomarkers including hsCRP, which, according to our findings, could mediate the lipid paradox. Similarly, another study conducted in 44,563 STEMI patients found that the “J-shaped” association between LDL-C and in-hospital mortality was attenuated after full adjustment for more than 40 covariates; however, this “J-shaped” association did not completely disappear [23]. Therefore, confounding factors could partly contribute to the lipid paradox, as patients with low LDL-C levels are more likely to be older, have more comorbidities, and receive more medications, but this cannot fully explain the phenomenon. Another explanation for the lipid paradox is inadequate nutritional status and frailty. A study by Lu et al. found that the lipid paradox was only observed in the population at high risk of malnutrition among AMI patients [25]. However, in the definition of nutritional risk, albumin plays a key role. Notably, albumin serves not only as a biomarker for nutritional status but also as an indicator of inflammation. Therefore, those defined as having high nutritional risk are also at high inflammatory risk.
The lipid paradox has been reported in other diseases characterized by inflammatory conditions. Several studies have revealed that hyperlipidemia was inversely associated with survival among hemodialysis patients, who often manifest a chronic inflammatory state [11, 26]. In a prospective study involving 823 dialysis patients, total cholesterol demonstrated a negative association with the risk of mortality in the presence of inflammation/malnutrition [indicated by serum albumin < 3.6 mg/dL, CRP ≥ 10 mg/L, or interleukin-6 (IL-6) ≥ 3.09 pg/mL], but showed a positive association with the risk of mortality in the absence of inflammation/malnutrition [11]. Similarly, lipid paradox has been observed in patients with rheumatoid arthritis, particularly in the presence of high levels of inflammation [27, 28]. Inflammation also plays a critical role in the pathogenesis of cardiovascular diseases, including acute coronary events. In AMI patients, inflammation may be triggered by cardiomyocyte death and can exacerbate the process of atherothrombosis [29]. HsCRP is widely considered the most representative biomarker of inflammation. Previous studies have shown that hsCRP tended to rise rapidly following AMI, and early elevation of hsCRP was associated with risks of recurrent cardiovascular events in patients with ACS [30, 31]. Our study found that the lipid paradox in AMI patients occurred predominantly in those with high hsCRP levels. This highlights the complex interplay between the lipid paradox and inflammation, particularly in diseases with high inflammatory burden.
Statins may play a key role in influencing the lipid paradox, as they not only lower LDL-C levels, but also reduce hsCRP levels. Clinical trials have shown that statins can decrease hsCRP levels independently of their effects on LDL-C [32, 33]. For instance, a study of 804,855 ischemic stroke patients found that the mediating effect of infection on the “U-shaped association” between LDL-C and mortality was more pronounced in patients without statin pretreatment [13]. Statins exert their anti-inflammatory effects on the vascular wall through molecular pathways in both the innate and adaptive immune systems, including the inhibition of the mevalonate pathway and isoprenoid formation [34]. In our study, the association between LDL-C and mortality risk was less pronounced in statin-treated patients, regardless of their inflammatory status. This effect was particularly evident in patients with high LDL-C levels, suggesting that statins primarily reduce mortality risk in this population through their lipid-lowering effects. Moreover, in the statin-pretreated group, patients with LDL-C levels ≥ 3 mmol/L exhibited a similar mortality risk. However, this result should be interpreted with caution. Residual confounding factors may contribute to these findings. Additionally, the relatively small proportion of patients with high LDL-C and hsCRP levels receiving statins in our cohort may limit the generalizability of these results. These aspects warrant further investigation to better understand the interplay between LDL-C, inflammation, and statin therapy in influencing mortality risk.
The mechanism underlying the interaction between inflammation and the lipid paradox in AMI is not well understood. LDL-C might be needed for the repairment of myocardial tissue, which could lead to a decrease in LDL-C levels [35]. It has been observed that the acute response to myocardial infarction can result in a decrease of LDL-C, which correlates with the infarct size [36, 37]. Furthermore, the elevation of hsCRP is induced by the myocardial damage during AMI. Thus, the relationship between LDL-C, hsCRP, and mortality risk may be influenced by the severity of AMI [38]. Additionally, the inflammation process itself can increase the cholesterol catabolism, engendering the increased expression of LDL receptors and oxidation of LDL [27]. Oxidized LDL, in turn, promotes the inflammation process, leading to further health deterioration and increased mortality risk [39].
However, some limitations in our study should be considered. First, inherent to the observational cohort design, despite adjusting for numerous covariates, the presence of potential unmeasured confounders and their interactions is inevitable. For instance, comorbidities may influence both inflammatory status and LDL-C levels, as well as the medical decisions and adherence to lipid-lowering medications, all of which could impact our results. Moreover, the impact of medical treatment during hospitalization could not be ignored. Consequently, our findings are exploratory and need validation through large clinical trials with longer follow-up periods.
Second, LDL-C and hsCRP levels were only measured at baseline, and the absence of follow-up laboratory data precluded the assessment of the impact of variations in LDL-C and hsCRP on mortality risk. However, it is worth noting that LDL-C and hsCRP measurements at different time points might provide additional insights. A Swedish nationwide cohort study found that LDL-C reduction 6 to 10 weeks after the index MI for patients with baseline LDL-C below 3.1 mmol/L—who face higher mortality risk due to the “lipid paradox” in this study—was associated with decreased all-cause mortality risk. This suggests that the lipid paradox may be independent of LDL-C changes and could diminish with different LDL-C measurements over time. Regarding hsCRP, hsCRP levels typically increase rapidly at presentation in ACS patients and return to baseline within weeks, indicating that hsCRP measured at this time reflects acute inflammation rather than ongoing status. Thus, hsCRP levels collected at different times may have distinct implications in the context of the lipid paradox.
Third, the relatively short follow-up period resulted in a limited event rate, potentially affecting the reliability of our estimates. However, our results indicate that the excessive mortality risk in patients with the lowest LDL-C quartile primarily occurred during the initial follow-up among those with high inflammatory status, suggesting that our findings would remain robust with extended follow-up.
Fourth, we only used hsCRP to define inflammatory status due to the limitation of our database. Other inflammatory biomarkers such as IL-6 and tumor necrosis factor-α (TNF-α) were not measured in our population. Both IL-6 and TNF-α can upregulate LDL receptor and scavenger receptor B1, leading to increased LDL uptake in the liver, which may help explain the lipid paradox [40, 41]. Cytokines such as IL-1 and TNF-α stimulate the production of IL-6, resulting in the synthesis of acute-phase reactants including CRP [42]. Therefore, given the role of CRP in the TNF-α/IL-6/CRP cascade, CRP serves as a reliable marker for reflecting the effects of IL-6 and TNF-α in this context. Future investigations should explore the consistency of TNF-α, IL-6, and CRP further.





Comments (0)