
Remember me






We systematically examined the distribution of ZNRF3 mutations in multiple human cancer types using cBioPortal (http://www.cbioportal.org/). As shown in Supplementary Fig. S1, truncating and missense variants appear evenly distributed along the ZNRF3 coding region, except for a lack of mutations in the first 100 amino acids encoded by exon 1. This latter exon is extremely GC-rich (Supplementary Fig. S2), which may lead to its frequent omission in next-generation sequencing analyses [12]. Notably, common germline ZNRF3 amino acid variants identified through the gnomAD database (https://gnomad.broadinstitute.org/) are not observed, indicating that basically all tumor-associated variants will be somatically acquired. Mutations are prevalent in uterus (5.8%) and bowel cancers (5%), with a predominance of missense mutations (Supplementary Fig. S3). Other tumor types with ZNRF3 mutation frequencies above 1% are those of the skin, liver, lungs, and cancers of the esophagus/stomach. Note that this analysis does not account for ZNRF3 deep deletions, which can also lead to ZNRF3 inactivation and are, for example, frequently observed in adrenocortical carcinomas [13].
The ZNRF3 long protein isoform is endogenously most relevantBoth a long ZNRF3 protein isoform (936aa) and shorter version (836aa) are reported in the Ensembl genome browser. The short isoform lacks the first 100 amino acids (Supplementary Fig. S4). To investigate which isoform is most relevant endogenously, we generated vectors expressing both ZNRF3 long and short protein isoforms. A β-catenin reporter assay showed that the ZNRF3 long isoform can significantly inhibit Wnt-induced β-catenin signaling in HEK293T cells, while this is not the case for the short variant (Supplementary Fig. S4). Next, we performed qPCR to test the expression of both ZNRF3 RNA variants in colorectal (n = 4), liver (n = 8) and pancreatic cancer (n = 4) cell lines. Although the short transcript can be detected in some cell lines, its expression is generally very low (Supplementary Fig. S5). Last, using signal peptide prediction shows that only the long isoform contains a genuine signal peptide at its N-terminus (Supplementary Fig. S6). Taken together, these observations show that only the ZNRF3 long isoform is clearly expressed and has the potential to transfer to the cell membrane to perform its principal function in reducing Wnt/β-catenin signaling.
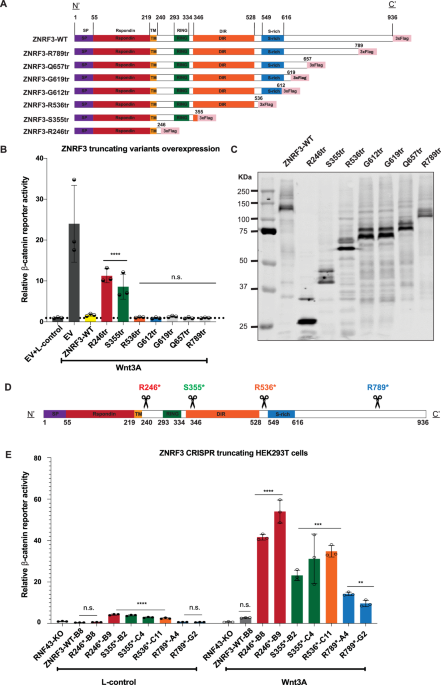
Analysis of ZNRF3 truncating mutationsVarious truncating ZNRF3 mutations have been observed in cancers (Supplementary Fig. S1), but thus far their functional consequences are largely unknown. Therefore, we generated expression vectors that can be grouped into two types of truncations (Fig. 1A). The first group delineates known functional domains (R246tr, S355tr, R536tr, R789tr), while the second set of truncations is potentially linked to a reported regulation of ZNRF3 by Caseine Kinase 1 (CK1). Both RNF43 and ZNRF3 are regulated in their function by CK1, which binds and phosphorylates residues in their S-rich domains (RNF43: ±455-507; ZNRF3: ±540-600) [14,15,16]. For RNF43 it was shown that truncating mutations in a region (D504-Q563) directly following this S-rich domain, lead to an aberrant Wnt-independent increase in β-catenin signaling by trapping CK1 at the plasma membrane [16]. Although ZNRF3 shows little homology with RNF43 in its C-terminal domain, we tested three tumor-associated truncating mutations reported in the equivalent ZNRF3 region (G612tr, G619tr, Q657tr).
Fig. 1: Characterization of ZNRF3 truncating mutations in regulating β-catenin signaling.
All tested ZNRF3 truncations lose some regulatory functions at the endogenous level, while the longer variants retain partial activity. A A schematic diagram depicting the 3xFLAG-tagged ZNRF3 truncating mutant expression vectors that were generated. For clarity, we refer to each domain by reported ZNRF3 functional domains, except for the CK1 binding domain, which overlaps with the S-rich domain but has not yet been identified accurately. B A β-catenin reporter assay in HEK293T cells shows that the variants retaining at least the DIR were able to inhibit Wnt-induced β-catenin signaling comparable to wild-type ZNRF3. Substantial and significant increases in signaling are observed in the shorter variants R246tr and S355tr. The relative Wnt β-catenin signaling activities are depicted as WRE/CMV-Renilla ratios, in which the value obtained for wild-type (WT) ZNRF3 was arbitrarily set to 1. Wnt3A conditioned medium was added to Empty Vector (EV) and all ZNRF3 variants transfected cells. L-control medium was added to EV transfected cells as a negative control for β-catenin signaling. C Immunoblot shows correct expression of the expected variants following HEK293T transfection. D A schematic overview of the ZNRF3 truncation variants generated in HEK293T-RNF43-knockout (KO) cells using CRISPR/Cas9-mediated gene editing. The scissors locations represent the CRISPR/Cas9 cutting sites where frameshift mutations were introduced. E RNF43-KO cells were used to exclude the influence of homologous RNF43. ZNRF3-WT-B8 cells represent an additional ZNRF3-WT control clone lacking RNF43, obtained during the gene-editing process. The relative β-catenin reporter activities are depicted as WRE/CMV-Renilla ratios, in which the value obtained for ZNRF3-WT-B8 clone treated with L-control medium, was arbitrarily set to 1. When exposed to L-control conditioned medium, all truncations except R246*-B8, R789*A4, and R789*G2, show some loss of β-catenin regulatory function when expressed at endogenous levels. In the Wnt3A conditioned medium, all truncations show some loss of β-catenin regulatory function, while the longer variants retain more function. All Dual-luciferase data are shown as mean ± SD. Statistical significance was assessed using a one-way ANOVA test (****P < 0.0001, ***P < 0.001, **P < 0.01).
When expressed transiently in HEK293T cells, all variants retaining at least the Disheveled interaction region (DIR) were able to inhibit Wnt-induced β-catenin signaling comparable to wild-type ZNRF3 (Fig. 1B, C). Shorter variants lost most of their ability to inhibit β-catenin signaling. Unlike RNF43, truncations of ZNRF3 directly following the S-rich CK1-regulatory domain did not lead to a dominant activation of β-catenin signaling [16].
These findings show that ZNRF3 and long variants thereof can effectively suppress β-catenin signaling when overexpressed. To test this at endogenous levels we generated CRISPR-Cas9-mediated HEK293T clones carrying ZNRF3 truncations (Fig. 1D, Supplementary Fig. S7). RNF43 knockout cells were used to exclude the effect of RNF43 inhibiting β-catenin signaling [7]. Two independent clones were obtained for each truncating variant in most cases. Interestingly, unlike the overexpression experiments, all truncations show some loss of β-catenin regulatory function when expressed at endogenous levels (Fig. 1E). Loss of functionality is inversely correlated to the length of the truncated ZNRF3 protein, with the longest R789tr protein retaining most functionality. Truncations following the RING domain (S355tr) or DIR domain (R536tr) show intermediate defects. Taken together, our results indicate that at endogenous levels all tested truncations lose some regulatory function, while the longer variants retain partial activity.
Identification of missense ZNRF3 variants affecting β-catenin signalingCurrently, all reported ZNRF3 missense variations are regarded as variants with uncertain significance (VUS), meaning that it is unknown whether they affect ZNRF3 protein function. In total 274 unique tumor-associated variants have been reported in cBioPortal until July 2023. To better understand which of these may act as driver mutations in tumorigenesis, we systematically analyzed patient-derived ZNRF3 missense mutants. Previously, we reported that RNF43 missense variants that affect its function are restricted to the RING and extracellular R-Spondin domains [8]. As these are also among the most homologous domains between RNF43 and ZNRF3 (Supplementary Fig. S8), we included all ZNRF3 variants in these domains reported at the time of our analysis. In addition, we included several variants identified at evolutionary conserved amino acids (Supplementary Fig. S9) reported in the transmembrane domain, the DIR and S-rich CK1-regulatory domain. The C-terminal domain of ZNRF3 (605–935) shows a low level of evolutionary conservation, making it unlikely that variants in this domain will affect ZNRF3 function.
Next, we tested all variants for their capability to inhibit Wnt-induced β-catenin signaling in HEK293T cells. In the R-Spondin-binding domain 12 out of 47 missense variants showed a significant loss-of-function (LOF) (Fig. 2A), even though for some specific variants the LOF was modest, e.g. G105C and A188V. Within the RING domain region 14 out of 27 tested missense variants showed a significant (partial) LOF or hyperactivating behavior (Fig. 3A). Among the 8 variants located at other domains only the D545N variant showed a significant but very modest loss of β-catenin regulatory function (Supplementary Fig. S10). In total, we identified 27 out of 82 tested variants that (partially) impaired ZNRF3 function.
Fig. 2: Analysis of missense variants within the ZNRF3 R-Spondin-binding domain.
Most variants in the ZNRF3 R-Spondin-binding domain with (partial) loss of function show reduced protein stability. A A β-catenin reporter assay reveals 12 variants that show a significant loss of β-catenin regulatory activity. Wnt3A conditioned medium was added to Empty Vector (EV) and all ZNRF3 variant transfected cells. L-control conditioned medium was added to EV transfected cells as a negative control for β-catenin signaling. All relative β-catenin reporter activities are depicted as WRE/CMV-Renilla ratios, in which the value obtained for ZNRF3-WT was arbitrarily set to 1. Statistical significance was assessed using a one-way ANOVA test (****P < 0.0001, ***P < 0.001, *P < 0.1). B All variants were co-transfected with an EGFP plasmid serving as transfection control. Using immunoblotting, 11/12 (partially) defective variants show a 3–9 fold reduction in protein stability, whereas most other variants retain normal or show a less reduced stability. All relative protein stabilities are depicted as ZNRF3/GFP ratios, in which the value obtained for ZNRF3-WT was set to 1.
Fig. 3: Analysis of missense variants within the ZNRF3 RING domain.
About half of the tested RING domain variants result in a (partial) loss-of-function or hyperactivating behavior of ZNRF3. A A β-catenin reporter assay reveals 14/27 variants that show a significant loss of β-catenin regulatory activity. Wnt3A conditioned medium was added to Empty Vector (EV) and all ZNRF3 variants transfected cells. L-control conditioned medium was added to EV transfected cells as a negative control for β-catenin signaling. All relative β-catenin reporter activities are depicted as WRE/CMV-Renilla ratios, in which the value obtained for ZNRF3-WT was arbitrarily set to 1 (****P < 0.0001). B Diagram depicting the RING domain structure of ZNRF3. The green circles are the residues required for interaction with ubiquitin-conjugating E2 enzymes, while yellow circles are the residues involved in Zinc (Zn) coordination. Tumor-associated variants that disrupt the β-catenin regulatory function of ZNRF3 are encircled by a red border. C All variants were co-transfected with EGFP plasmids. Immunoblot shows that all defective variants show at least a 1.5-fold increased protein stability, with the exception of E305K and D321N. All relative protein stabilities are depicted as ZNRF3 variants/GFP ratios, in which the value obtained from ZNRF3-WT was set to 1.
Focusing on the RING domain, as expected mutations of the C/H residues essential for Zinc interaction and formation of the RING structure, all disrupt β-catenin regulation (Fig. 3B). The same holds true for mutants observed at residues required for interaction with ubiquitin conjugating E2 enzymes (green circles in Fig. 3B) [17], except for the I295M variant where one hydrophobic residue is replaced by another. Next, we made a comparison with reported variants of the homologous RING structure of RNF43 [8, 18], which we extended by investigating an additional 28 previously unexplored variants (Supplementary Fig. S11). In total, 39 out of 44 RING domain variants disrupt RNF43 function, which is a significantly higher proportion than observed in the ZNRF3 RING domain structure (P < 0.001, Chi-square test). Supplementary Fig. S12 provides schematic representations of the RING structures of RNF43/ZNRF3 and location of variants disrupting their function.
ZNRF3 variants defective in β-catenin regulation affect protein stabilityTo better define the mechanism through which LOF/hyperactivating ZNRF3 variants lose β-catenin regulatory activity, we first determined their protein stability. Basically all ZNRF3/RNF43 RING domain variants defective in β-catenin regulation, resulted in increased protein levels (Fig. 3C, Supplementary Fig. S11B), which is not unexpected given that these variants will also have lost their auto-ubiquitination potential [9, 10, 19].
The opposite effect was observed for 11 out of 12 R-Spondin domain variants affecting β-catenin signaling (Fig. 2B), which showed a clear 3–9 fold reduction in overall protein levels compared with wild-type ZNRF3. Similar observations were made for R-Spondin domain variants within the RNF43 protein, but less consistently (Supplementary Fig. S13). While several defective RNF43 variants show a 3–15 fold reduction in protein levels (A46V, I48T, P52L, G80R, P154L), others show a more modest about twofold reduction (G67C, A73F, A73V, A136T, P160S, A169T) or levels comparable to wild-type (K108E, G166C). Conversely, while most variants retaining normal β-catenin regulation in overexpression studies show protein levels equal to or at most twofold reduced compared to wild-type, the A78T variant is unstable despite retaining β-catenin regulation. These results show that most defective R-Spondin domain variants of ZNRF3 are reduced in protein stability, while this is the case for several but not all RNF43 variants.
LOF missense mutants in ZNRF3 R-Spondin domain fail to reach the membrane correctlyWe used a cell surface biotinylation assay to determine if the defective missense ZNRF3 variants can reach the plasma membrane correctly (Fig. 4A). As shown in Fig. 4B, all tested RING domain variants are clearly present at the membrane. Variants that do not impair ZNRF3 functionality (E304D and R314Q) are detectable at levels comparable to wild-type ZNRF3, while defective variants are detected at increased levels, consistent with their increased stability. In contrast, defective variants within the R-Spondin-binding domain are barely detectable using this assay, indicating that they are strongly impaired in being transferred from the endoplasmic reticulum (ER) to the plasma membrane.
Fig. 4: LOF missense mutants in ZNRF3 R-Spondin domain fail to reach the membrane correctly.
Analysis of plasma membrane presence of selected R-Spondin and RING domain ZNRF3 variants. A Schematic overview showing the process of biotinylation and IP experiments. The Sulfo-NHS-LC-Biotin reagent is added to live cells for cell membrane protein labeling, followed by α-FLAG beads immunoprecipitation of ZNRF3/RNF43 FLAG-tagged protein. Next, IRDye800CW-streptavidin is used to label Biotin to measure the protein levels of ZNRF3/RNF43 reaching the cell membrane. B In contrast to wild-type ZNRF3 and non-defective R-Spondin domain variants, six tested variants defective in β-catenin regulation (red squares) fail to reach the membrane correctly. Defective RING domain variants (I295T to C333S) have no problems in reaching the membrane and are detected at increased levels in line with their increased overall stability. C HeLa cells were transfected with ZNRF3 variants. Expression and distribution of ZNRF3 variants was assessed by immunofluorescence. The signal of MYC (red) indicates the ZNRF3 expression on the cell membrane, and the signal of HA (green) indicates the ZNRF3 expression on cell membrane or in cytoplasm. The ZNRF3 WT and R149Q variants are capable of reaching the cell membrane, while G150V, P179L, and I205T, which lead to the loss of function of ZNRF3, cannot be detected at the cell membrane.
We confirmed this observation by performing live cell staining of 3xMyc-ZNRF3-HA transfected HeLa cells (Supplementary Fig. S14A). The experimental procedure involved incubating the cells at 4 °C, followed by exposure to anti-Myc-tag antibodies (visualized in red). Next, cells were fixed, permeabilized and additionally stained for the intracellular HA-tag (visualized in green). Using this approach, we will only obtain Myc-tag signal when ZNRF3 can efficiently reach the membrane, which was readily observed for wild-type ZNRF3 and the R149Q variant that is not defective in β-catenin regulation (Fig. 4C, Supplementary Fig. S14B). In contrast, the three defective variants (G150V, P179L, and I205T) did not yield any signal for the Myc-tag, while they were efficiently detected using the intracellular HA-tag. We also applied this method to RNF43 and R-Spondin domain defective variants thereof. As expected, wild-type RNF43 exhibited a clear Myc-tag staining at the membrane, whereas the missense variants exhibited a noticeably reduced but not entirely absent staining (Supplementary Fig. S15). Taken together, these findings show that R-Spondin/PA domain missense variants of RNF43 and ZNRF3 do not or less efficiently localize to the membrane.
Comparing R-Spondin/PA domain deletions with defective missense mutationsPreviously, several reports showed that deleting large portions of the R-Spondin interaction domains of RNF43 and ZNRF3, results in proteins that can regulate Wnt-induced β-catenin signaling as efficiently as their wild-type counterparts [20,21,22,23]. We confirmed this observation for three different R-Spondin domain ZNRF3 deletions (Fig. 5A). This leads to the apparent contradiction that specific missense mutations in this domain are more detrimental to ZNRF3 function than large deletions. To explore this discrepancy, we investigated whether these deletion variants properly localize to the cell membrane. Using the previously described immunofluorescence method, we observed that all deletion variants were detectable at the membrane, similar to wild-type ZNRF3 (Fig. 5C). Additionally, western blot analysis showed a comparable (Δ57-207; Δ103-205) or slightly reduced stability (Δ101-142) for these deletion variants (Fig. 5B). Thus, in contrast to specific missense variants, these deletion mutants retain the ability to correctly traffic to the membrane and regulate Wnt-receptor levels.
Fig. 5: ZNRF3 R-Spondin binding domain deletions retain the ability to correctly traffic to the membrane and regulate Wnt-induced β-catenin signaling.
Comparing R-Spondin/PA domain deletions with defective missense mutations. A A β-catenin reporter assay reveals that LOF missense R-spondin domain variants are defective in reducing Wnt-induced β-catenin signaling. However, ZNRF3 with deletions in the same R-spondin binding domain still behave similar to WT. Wnt3A conditioned medium was added to Empty Vector (EV) and all ZNRF3 variant transfected cells. L-control conditioned medium was added to EV transfected cells as a negative control for β-catenin signaling. All relative β-catenin reporter activities are depicted as WRE/CMV-Renilla ratios, in which the value obtained for ZNRF3-WT was arbitrarily set to 1. Statistical significance was assessed using a t-test (*P < 0.1, **P < 0.01). B ZNRF3 variants were co-transfected with an EGFP plasmid serving as transfection control. Immunoblot shows that Δ57-207 and Δ103-205 exhibit comparable expression to WT, and Δ101-142 shows slightly reduced stability. The missense variants show strong protein instability. All relative protein stabilities are depicted as ZNRF3 variants/GFP ratios, in which the value obtained from ZNRF3-WT was set to 1. C HeLa cells were transfected with ZNRF3 variants. Expression and distribution of ZNRF3 variants were assessed by immunofluorescence. The signal of MYC (red) indicates the ZNRF3 expression on the cell membrane, and the signal of HA (green) indicates the ZNRF3 expression on cell membrane or in cytoplasm. ZNRF3 WT and all R-Spondin deletions are capable of efficiently reaching the cell membrane.
Low temperature partially rescues missense mutations in the R-spondin-binding domainBoth the reduced protein stability of R-Spondin domain variants as well as their failure to reach the membrane, are indicative of a problem with protein misfolding. Within the endoplasmic reticulum, the structures of newly synthesized proteins undergo a rigorous quality control and when improperly folded are removed by ER-associated degradation [24]. Similar observations have been made for other cell surface located proteins. One well-known example is the common F508del mutation in CFTR, leading to cystic fibrosis. The main consequence of this mutation is a defect in protein folding and trafficking, which can be rescued by growing cells at reduced temperatures, which has also been observed for other proteins [25, 26]. Therefore, we tested if the membrane location of R-Spondin variants can be restored by culturing cells at 27 °C. We tested two variants for both RNF43 and ZNRF3 that all show reduced membrane presence at 37 °C. In all four cases surface biotinylation was increased when cultured at lower temperature, although not reaching the levels of the wild-type proteins (Fig. 6A). Next, using a β-catenin reporter assay we determined if low culture temperatures can also significantly affect β-catenin regulation. Out of 12 tested ZNRF3 variants, 7 showed a near-complete restoration of their regulatory function, while this was only observed for 2 out of 14 RNF43 variants (Fig. 6B/C).
Fig. 6: Low temperature partially rescues missense mutations in the R-Spondin-binding domain.
Culturing cells at 27 °C supports variants of the ZNRF3 R-Spondin-binding domain to (partially) regain their function in regulating β-catenin signaling. A Left panel shows an IP-experiment of surface-biotinylated RNF43 and ZNRF3 R-Spondin domain mutants cultured at 37 °C or 27 °C. Mutants fail to reach the cell membrane when cultured at 37 °C, while they show improved membrane presence when grown at 27 °C. B An assessment of β-catenin reporter activity shows that 7/12 variants in the ZNRF3 R-Spondin-binding domain regain functionality in Wnt/β-catenin signaling regulation when cultured at 27 °C. C The same analysis performed for 14 RNF43 variants shows that only the G166C and A169T variants can be rescued at 27 °C. Wnt3A conditioned medium was added to Empty Vector (EV) and all RNF43/ZNRF3 variants transfected cells. L-control conditioned medium was added to EV-transfected cells as a negative control for β-catenin signaling. All relative β-catenin reporter activities are depicted as WRE/CMV-Renilla ratios, in which the value obtained for RNF43-WT or ZNRF3-WT was arbitrarily set to 1. Statistical significance was assessed using a one-way ANOVA test (****P < 0.0001, ***P < 0.001, *P < 0.1).
Taken together, our results indicate that the main functional consequence of R-Spondin domain mutants of especially ZNRF3, appears to be a failure to fold correctly, leading to reduced protein stability and trafficking to the membrane. These defects are temperature-sensitive as they can partially be rescued by lower temperatures.
Testing “dominant-negative” potential of R-Spondin/RING variants at endogenous levelsAlready in the first papers linking RNF43 and ZNRF3 to Wnt/β-catenin signaling, it was noted that mutating their RING domains led to hyperactivation of β-catenin signaling, indicating a dominant-negative mode of action [9, 10]. In subsequent years this observation was confirmed in several publications and also extended to mutations in the R-Spondin domain [8, 11, 15, 18]. Here, we show the same for specific ZNRF3 variants (e.g. P179L, P331H). The underlying mechanism relies on the ability of RNF43 and ZNRF3 to form homo- and heterodimers [8, 27]. It is believed that if one of the dimer-partners carries an inactivating RING or R-Spondin mutation, it can render the entire dimer inactive in a dominant-negative manner (Supplementary Fig. S17A). This would explain why transient overexpression of such variants leads to hyperactivation, as it will also inhibit endogenously expressed RNF43/ZNRF3 [8]. This observation has led to the speculation that these missense variants may have oncogenic potential [8], meaning that one such RNF43/ZNRF3 mutation may already sufficiently increase β-catenin signaling to drive tumorigenesis, and that no second hit is required.
Thus far this speculation is entirely based on overexpression experiments, which, however, leads to a more than 1000-fold overexpression on RNA level (Supplementary Fig. S16A), and most likely also on protein level. Therefore, we decided to test the relevance of “dominant-negative” variants at endogenous levels. Using gene-editing we generated various HEK293T clones expressing R-Spondin (P179L) or RING domain ZNRF3 variants (R307W, P311L/H) in a heterozygous fashion. On purpose, we used wild-type HEK293T cells to determine if the remaining wild-type RNF43 and ZNRF3 can be sufficiently inhibited. HEK293T cells express ZNRF3 at approximately 20–30-fold higher levels than RNF43 (Supplementary Fig. S16B) [9], indicating that expression levels of mutant ZNRF3 are likely sufficiently high to impose a dominant-negative effect on wild-type RNF43/ZNRF3. Importantly, we also generated four clones carrying a heterozygous ZNRF3 knockout, to allow a direct comparison between loss-of-function and supposed dominant-negative mutations.
Next, we exposed all clones to exogenous Wnt3A and determined β-catenin reporter activity (Fig. 7A, Supplementary Fig. S17B). Although we do observe some clone-to-clone variation, basically all heterozygous mutant clones show a modest 2–8 fold activation of β-catenin signaling compared to wild-type cells. Importantly, we see no clear difference in activation when comparing heterozygous knock-out of ZNRF3 (lane 5) with clones heterozygously expressing “hyperactive” variants of ZNRF3 (lanes 2–4), indicating that the proposed dominant-negative behavior does not exist or is very weak. The low level induction of β-catenin signaling in all heterozygous clones, also suggests that partial loss of ZNRF3 expression leads to haploinsufficiency, at least in HEK293T cells.
Fig. 7: Testing dominant-negative potential of ZNRF3 variants at endogenous levels.
A Endogenous levels of heterozygous missense mutations do not affect ZNRF3 function more strongly than heterozygous knockout mutations. β-catenin reporter assays for knockin/out ZNRF3 clones. All heterozygous mutations, including P179L, R307W, P331L/H, and WT+frameshift (fs), slightly increase β-catenin signaling (2–8 fold) to similar levels. Homozygous ZNRF3 knockout clones, homozygous R307W or P331H knockin clones, and RNF43/ZNRF3 double knockout clones, all show a comparable further increase in signaling (18–30 fold). The relative Wnt/β-catenin signaling activities are depicted as WRE/CMV-Renilla ratios, in which the value obtained for wild-type (WT) HEK293T cells was arbitrarily set to 1. Wnt3A conditioned medium was added to WT HEK293T cells and all CRISPR-edited HEK293T cells. Statistical significance was assessed using a T-test (****P < 0.0001, **P < 0.01, *P < 0.1). For each heterozygous amino acid variant, 3–4 individual clones were used. Values for each individual clone are shown in Supplementary Fig. 17B. B A β-catenin reporter activity analysis performed for representative supposedly dominant-negative RNF43 or ZNRF3 variants, shows that overexpression of these hyperactive variants reduces β-catenin signaling much weaker than a complete knockout of both proteins. Wnt3A was added to all samples except for control-treated empty vector (EV) transfected cells, serving as negative control. All relative β-catenin reporter activities are depicted as WRE/CMV-Renilla ratios, in which the value obtained for EV with L-Control was arbitrarily set to 1. Statistical significance was assessed using a one-way ANOVA test (****P < 0.0001).
We also obtained clones with homozygous expression of R307W and P331H variants, both of which exhibit increased β-catenin reporter activity compared to heterozygous clones (Fig. 7A, Supplementary Fig. S17B). However, in a direct comparison with previously generated ZNRF3- and double RNF43/ZNRF3 knockout clones, no significant difference is observed.
To further demonstrate that the proposed dominant-negative behavior of hyperactive variants is rather weak, we performed a direct comparison of β-catenin reporter activity observed in double RNF43/ZNRF3 knockout clones and those of overexpressed hyperactive variants. If overexpression of these variants would be very effective in blocking endogenous RNF43/ZNRF3, it would result in a comparable activation of β-catenin signaling. However, induction of β-catenin signaling by selected RNF43 and ZNRF3 hyperactive variants is much lower compared with a complete knockout of both proteins (Fig. 7B).
To summarize, at endogenous expression levels all tested RING and R-Spondin ZNRF3 domain variants, do not possess sufficient dominant-negative activity to impair the function of the remaining wild-type RNF43 and ZNRF3 proteins. In other words, they appear to behave as classical loss-of-function mutations.
ZNRF3 mutations often co-occur with other β-catenin activating gene mutations in colorectal cancerThus far our data show that ZNRF3 truncating mutations can be regarded as (partial) loss of function mutations, while this applies to a selected number of missense mutations. We explored the cBioPortal database to identify which cancers carry these defective ZNRF3 variants (Supplementary Table 1). They are identified at low numbers in various cancer types, such as those of the endometrium, stomach, liver and adrenal gland, but are most frequently observed in colorectal cancers (63 in about 2100 CRC patients profiled for ZNRF3, i.e. ± 3%). As shown in Fig. 8 and Supplementary Table 1, CRCs with defective ZNRF3 mutations are highly enriched for BRAF-V600E mutation, a mismatch-repair defect, and right-sided tumor location (41/63, 46/56, and 39/47, respectively). Next, we determined to what extent ZNRF3 mutations are accompanied by other gene mutations that result in β-catenin activation (Fig. 8, Supplementary Table 1). Out of 63 cancers, we identified 12 that apparently only carry inactivating ZNRF3 mutations. ZNRF3-mutant CRCs carrying additional APC truncations or oncogenic CTNNB1 mutations classically linked to β-catenin activation, are only observed in 5 and 1 cancer, respectively. The remaining 45 cancers carry additional truncating mutations in either AXIN1, AXIN2 or AMER1, or defective missense or truncating RNF43 mutations [7, 8, 28,
Comments (0)