
Remember me
To elucidate the potential role of USP36 in the progression of CRC, a significant upregulation of USP36 was initially identified in CRC tissues in the TCGA database (Fig. S1A). Meanwhile, analysis of public databases including GEO (GSE100179, GSE41258) and public gene chip data, revealed that USP36 expression levels were increased in adenoma, primary tumor, and metastasis, relative to adjacent normal tissue (Fig. S1B, C). Moreover, Kaplan-Meier analysis of the GEO database (GSE40967) indicated that patients with higher USP36 expression exhibited reduced relapse-free survival and overall survival rates (Fig. S1D). Next, gene set enrichment analysis (GSEA) in TCGA datasets was conducted to explore the CRC-related signaling pathway regulated by USP36. As shown in Fig. 1A, enrichment plots demonstrated that USP36 was negatively correlated with p53 signaling pathway. Subsequently, we further investigated whether USP36 modulates p53 signaling pathway activation.
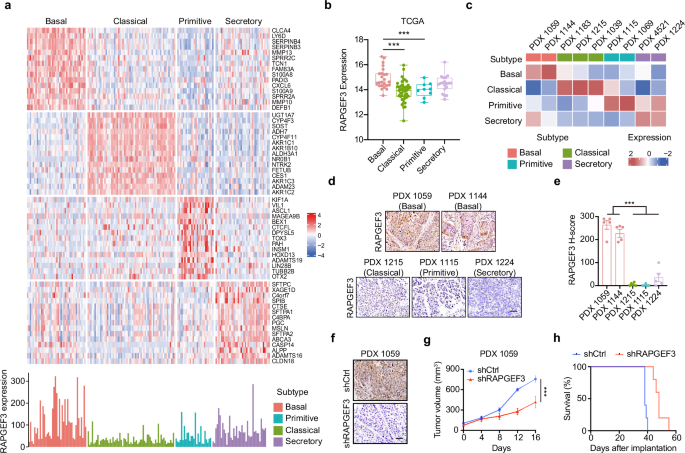
Fig. 1: USP36 inhibits p53 signaling pathway by impairing p53 transcriptional activity.
A GSEA demonstrates the correlation between USP36 and p53 signaling pathway in the enrichment plot. Data were obtained from the TCGA database. NES normalized enrichment score. FDR false discovery rate. B, C Relative mRNA and protein level of p53 and target genes in CRC cells upon USP36 knockdown or overexpression. D Luciferase reporter assays indicated shUSP36 markedly activated p53 signaling and p53 inhibitor (pifithrin-α) restored the phenotype (left panel). USP36-induced inhibition of p53 transcriptional activity was reversed by p53 overexpression (right panel). E, F ChIP-qPCR assay was performed to detect the binding ability of p53 to the CDKN1A or BAX promoter in CRC cells with USP36 knockdown or overexpression. All data are presented as the means ± SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ns not significant.
We established the shCtrl, shUSP36 #1/#2/#3, vector, and USP36 plasmids to construct stable cell lines with lentivirus-mediated transfection. Based on the efficiency validation results, shUSP36 #1 and shUSP36 #3 were preferred for further experiments owing to their superior knockdown efficacy compared to shUSP36 #2 (Figs. 5A, B and S3A). Then, we observed that USP36 depletion in HCT 116 and RKO cells upregulated, while USP36 overexpression in LoVo cells decreased, the mRNA and protein expression of p53 target genes (Figs. 1B, C and S1E). However, the mRNA and protein levels of p53 remained unchanged with alternations of USP36 in CRC cells. Further, luciferase reporter assays indicated that p53 transcriptional activity was augmented by USP36 silencing in HCT 116 and RKO cells, which was restored by pifithrin-α (PFT-α, a p53-specific inhibitor) (Figs. 1D and S1F). In contrast, USP36 overexpression-mediated inhibition of p53 transcriptional activity was reversed by p53 overexpression in LoVo cells (Fig. 1D). Chromatin immunoprecipitation (ChIP) assays also showed that USP36 knockdown enhanced the ability of p53 to bind to the promoters of its canonical downstream genes, such as p21 and BAX, whereas USP36 overexpression exhibited an opposite effect (Fig. 1E, F). Collectively, these results indicated that USP36 inhibits p53 transcriptional activity and negatively regulates p53 signaling pathway.
USP36 interacts with and upregulates RBM28 depending on its deubiquitinase activityTo further determine the underlying mechanism of USP36-mediated inhibition of p53 signaling pathway, immunoprecipitation‐based mass spectrometry (IP‐MS) was performed to identify USP36-interacting proteins. Of note, among the top 10 proteins identified in the USP36 complex (Fig. S2A), we found RBM28 had the highest abundance which had also been previously reported to negatively regulate p53 transcriptional activity. Therefore, we speculated that RBM28 could be a potential target for USP36 to regulate p53 signaling pathway. Immunofluorescence (IF) assay exhibited the co-localization of USP36 and RBM28 in the nucleus in the HCT 116 and HEK293T cells (Fig. 2A). To verify the physical interaction between USP36 and RBM28, co-immunoprecipitation (Co-IP) were performed in HEK293T and CRC cells. The results revealed that among Flag-tagged USP36 immunoprecipitates detected Myc-tagged RBM28 and vice versa (Fig. 2B). More importantly, endogenous interactions between USP36 and RBM28 were validated in CRC cells (Fig. 2C). Moreover, GST-pulldown assay revealed that USP36 directly interacted with RBM28 in vitro (Fig. 2D). Then, truncated mutant fragments were generated to determine the specific regions responsible for USP36 and RBM28 interaction. Immunoprecipitation assay revealed that USP36 interacted with RBM28 through its USP domain (Fig. 2E), whereas the RRM2 domain of RBM28 mediated the interaction with USP36 (Fig. 2F). These findings suggest an interaction between USP36 and RBM28.
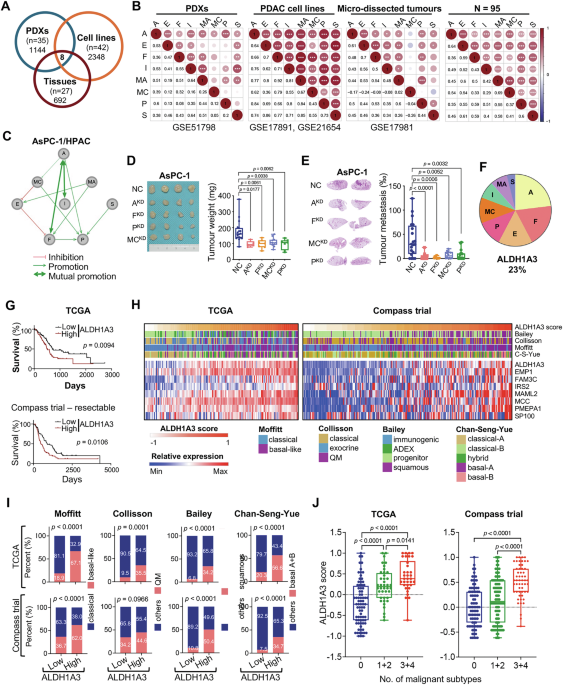
Fig. 2: USP36 interacts with and upregulates RBM28 depending on its deubiquitinase activity.
A HCT 116 and HEK293T cells were fixed and stained with USP36 antibody (Red) and RBM28 antibody (Green). Nuclei were stained with DAPI (blue). Scale bars, 10 µm. B HEK293T cells transfected with Flag-USP36 and Myc-RBM28 were harvested to validate the exogenous interaction between USP36 and RBM28 by Co-IP and western blot. C Cell lysates from HCT 116, RKO, and LoVo cells were immunoprecipitated with IgG or an anti-USP36 and anti-RBM28 antibody, followed by western blotting. D Lysates from HEK293T cells transfected with Myc-RBM28 were incubated with GST‐USP36 or GST protein. The interacted RBM28 was detected by western blot. E Schematic representation of USP36 domain and truncations (upper panel). HEK293T cells were cotransfected with Myc-RBM28 and Flag-USP36 or its mutants. The cell lysates were analyzed by IP with an anti-Flag antibody (lower panel). F Schematic representation of RBM28 domain and truncations (upper panel). HEK293T cells were cotransfected with Flag-USP36 and Myc-RBM28 or its mutants. The cell lysates were analyzed by IP with an anti-Myc antibody followed by western bolting (lower panel). G Western blotting was used to detect RBM28 protein levels in HCT116 and RKO cells transfected with the indicated shRNA. H Western blotting was employed to assess RBM28 protein levels in LoVo cells transfected with wild-type USP36 or USP36 C131A. I LoVo cells were transfected with increasing amounts of USP36WT or USP36C131A for 48 h and cell lysates were analyzed by western blot.
Next, we proceeded to investigate whether the expression levels of RBM28 are modulated by USP36. It was observed that the depletion of USP36 led to a decrease in RBM28 protein levels in both HCT 116 and RKO cells (Fig. 2G). Consistently, wild-type(WT) USP36 significantly increased RBM28 protein levels in a dose-dependent manner, whereas catalytic inactive mutant C131A (USP36C131A) showed no obvious impact on RBM28 protein levels (Fig. 2H, I). However, there were no observable changes in the mRNA levels of RBM28 following USP36 knockdown or overexpression, which suggested that USP36 upregulates RBM28 expression through post-translational regulation (Fig. S2B). These results indicate that USP36 regulates RBM28 expression in a DUB activity-dependent manner.
USP36 stabilizes and deubiquitylates RBM28 at K162 residueTo further investigate whether USP36 regulates RBM28 expression through the proteasome pathway, We exposed CRC cells with USP36 knockdown to the proteasome inhibitor MG132. As shown in Fig. 3A, MG132 could reverse RBM28 protein levels caused by USP36 knockdown. In addition, Cycloheximide (CHX) chase assay showed that USP36 knockdown accelerated the degradation of RBM28 (Figs. 3B and S2C, D). In contrast, the ectopic overexpression of USP36WT, but not USP36C131A, prolonged the half-life of RBM28 (Fig. 3B). These results indicate that USP36 stabilizes RBM28 through the prevention of the proteasomal degradation pathway.
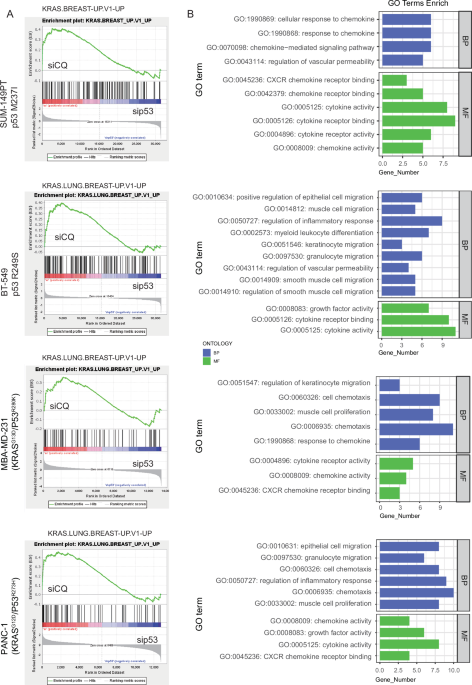
Fig. 3: USP36 stabilizes and deubiquitylates RBM28 at K162 residue.
A HCT 116 and RKO cells with knockdown of USP36 were treated with or without the proteasome inhibitor MG132 (10 µM, 6 h), and then proteins were analyzed. B Stability analysis of RBM28 protein in HCT 116-shUSP36 #1/3 cells, LoVo-USP36WT, LoVo-USP36C131A cells and treated with 10 µg/mL cycloheximide (CHX) for indicated times. The lower panels are quantification of RBM28 protein levels. C HCT 116 and RKO cells depleted with USP36 were treated with 10 μM of MG132 for 6 h and then harvested. RBM28 was immunoprecipitated with anti‐RBM28 and immunoblotted with anti‐HA. D LoVo cells transfected with the indicated plasmids were treated with 10 μM of MG132 for 6 h and subjected to the sequential IP and immunoblotting analysis. E Immunoblotting was used to detect the ubiquitination of RBM28 in HEK293T cells co‐transfected with Myc‐RBM28, HA‐Ubiquitin, and Flag‐USP36 (wild‐type or C131A). Cells were treated with 10 μM MG132 for 6 h before harvesting. F USP36 removed the ubiquitin chain of RBM28 in a time- and dose-dependent manner. G Immunoblotting to detect the ubiquitination of RBM28 mutants in HEK293T cells co‐transfected with Myc‐RBM28 mutants, Flag-USP36, and HA‐Ub. Cells were treated with 10 μM MG132 for 6 h before harvesting. H HEK293T cells were transfected with Myc-RBM28-WT or Myc-RBM28-K162R plasmids and treated with 10 µg/mL CHX for the indicated time and then were analyzed by western blot. Quantification of the expression levels of WT and K162R RBM28 was shown in the lower panel. All data are presented as the means ± SD of three independent experiments. ***p < 0.001.
Since USP36 is a member of the deubiquitinase family, we explored whether USP36 deubiquitylates RBM28. USP36 depletion markedly increased endogenous RBM28 ubiquitination in HCT 116 and RKO cells (Fig. 3C). Conversely, ectopic overexpression of USP36WT, but not USP36C131A, dramatically decreased the ubiquitination level of RBM28 in LoVo and HEK293T (Fig. 3D, E). In vivo ubiquitination assays also demonstrated that USP36 removed the ubiquitin chain of RBM28 in a time- and dose-dependent manner (Fig. 3F). Considering the prevalence of K29, K48, and K63 chains in regulating protein stability, we explored the specific ubiquitin chain responsible for RBM28 degradation. Deubiquitination assays showed that USP36 predominantly removed K48-linked ubiquitin chains from RBM28 (Fig. S2E).
To further explore the specific sites of USP36-mediated deubiquitination on RBM28, a total of 5 potential lysine residues were predicted using bioinformatic tools (https://www.phosphosite.org). Next, we constructed point mutant plasmids of RBM28 by substituting lysine residues with arginine residues. As shown in Fig. 3G, we found that USP36 did not regulate the ubiquitination levels of K162R mutants, whereas the ubiquitination levels of wild-type RBM28 and other mutants (K39R, K172R, K708R, K738R) exhibited a significant decrease. Additionally, the protein half-life of the K162R mutant is extended relative to wild-type RBM28 (Fig. 3H). These findings demonstrate that USP36 stabilizes RBM28 through deubiquitination at K162 residue.
RBM28 is required for USP36-mediated inhibition of p53 transcriptional activityIt has been previously reported that RBM28 binds to the DNA-binding region of p53, leading to the suppression of p53-mediated transcription [26]. Considering the stabilizing effect of USP36 on RBM28, we hypothesized whether USP36 inactivates p53 signaling pathway via RBM28. As expected, GSEA in the TCGA database indicated that RBM28 negatively correlated with p53 signaling pathway in CRC (Figs. 4A and S2F). Co-IP experiments suggested that RBM28 could interact with p53 in CRC cells (Fig. 4B). Additionally, the nuclear co-localization of RBM28 and p53 in CRC cells was evidenced by IF staining assay (Fig. 4C). Then, we found that RBM28 overexpression led to a notable decrease in the mRNA and protein levels of p53 target genes, while its knockdown yielded contrary results (Figs. 4D, E and S2H, I). Luciferase reporter assay demonstrated a decrease in p53 transcriptional activity upon RBM28 overexpression, which was enhanced by RBM28 knockdown (Figs. 4F and S2J). ChIP assays revealed that overexpressed RBM28 suppressed the capacity of p53 to bind to the promoters of indicated genes, while depleted RBM28 showed a converse outcome (Figs. 4G, H and S2K). Together, these findings illustrated that RBM28 binds to p53, resulting in the inhibition of its transcriptional activity.
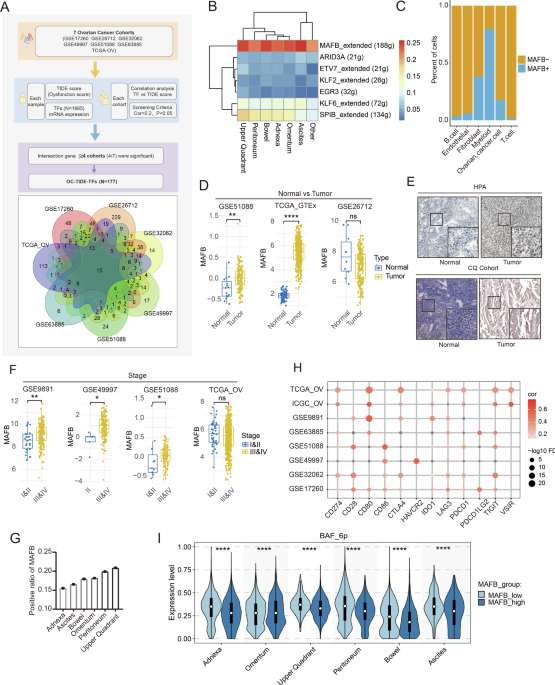
Fig. 4: RBM28 is required for USP36-mediated inhibition of p53 transcriptional activity.
A GSEA demonstrates the correlation between RBM28 and p53 signaling pathway in the enrichment plot. Data were obtained from the TCGA database. NES normalized enrichment score. FDR false discovery rate. B Co‐IP assay reveals an association between endogenous RBM28 and p53 in CRC cells. Lysates from HCT 116 cells, RKO cells, and LoVo cells were immunoprecipitated with control IgG or an anti-p53 and anti-RBM28 antibody, followed by immunoblotting analysis. Lysate of the indicated CRC cells was used as the input. C IF assays displayed subcellular co-localization of p53 and RBM28 in HCT 116 cells. HCT 116 cells were fixed and stained with RBM28 antibody (Green) and p53 antibody (Red). Nuclei were stained with DAPI (blue). Scale bars, 10 µm. D, E HCT 116-shUSP36 or LoVo-overexpressed USP36 cells were respectively treated with RBM28 lentivirus transfection (left panel) or shRBM28 (right panel), in which the p53 and target genes were detected by qRT-PCR and western blot. F Luciferase reporter assays were performed to detect the activation of p53 signaling in the indicated cells. G, H The binding ability of p53 to the CDKN1A or BAX promoter in the indicated cells was evaluated by ChIP-qPCR assay. All data are presented as the means ± SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ns not significant.
Meanwhile, we observed that USP36 depletion-mediated upregulation of p53 target genes was reversed by RBM28 overexpression, and vice versa (Fig. 4D, E). In addition, overexpression of RBM28 restored the luciferase activity enhanced by knockdown of USP36, whereas RBM28 knockdown showed the opposite effect (Figs. 4F and S2J). Overexpression of RBM28 also reversed the increased binding of p53 to the promoter of its target genes induced by USP36 knockdown, while the opposite results were observed upon RBM28 knockdown (Figs. 4G, H and S2K). Based on these results, RBM28 acts as an essential mediator for p53 signaling pathway regulated by USP36.
USP36 promotes CRC cell proliferation, migration, and invasion in vitroGiven the stabilizing impact of USP36 on RBM28-mediated p53 inhibition and the function of p53 in tumorigenesis, we evaluated the biological role of USP36 in CRC. Cell proliferation was evaluated through CCK8 and colony formation assays. The findings indicated a significant inhibition of cell proliferation in HCT 116 and RKO cells upon USP36 silencing, whereas its overexpression in LoVo cells exerted the opposite function (Figs. 5C–F and S3B, C). Furthermore, flow cytometry apoptosis assays revealed an increase in the proportion of apoptotic cells following USP36 knockdown of HCT 116 and RKO cells, while overexpression resulted in a reduction in apoptosis in LoVo cells (Figs. 5G, H and S3D). Subsequently, USP36 silencing was shown to suppress migration and invasion in HCT 116 and RKO cells, as evidenced by transwell and wound healing assays, while its overexpression led to opposite outcomes (Figs. 5I–L and S3E, F). Collectively, in vitro experiments indicate that USP36 could stimulate the growth of CRC cells and enhance their migratory and invasive capabilities.
Fig. 5: USP36 promotes CRC proliferation and metastasis in vitro.
A, B Transfection efficiency of USP36 in HCT 116 and LoVo cells, detected by qRT-PCR and WB. C, D CCK8 assays were applied to detect the viability of USP36 knockdown or overexpression cells. E, F Colony formation assays were used to evaluate the proliferation of CRC cells. G, H Flow cytometry was used to analyze the apoptosis rates (LR + UR) of cells. LR, early apoptotic cells; UR, terminal apoptotic cells. I, J Wound healing assays were used to assess cell migration ability. K, L Transwell assays were performed to evaluate the migration and invasion abilities of CRC cells. All data are presented as the means ± SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
USP36 facilitates CRC proliferation and metastasis in vivoTo further explore the in vivo functions of USP36, CRC cells mentioned above were conducted to establish xenograft tumor and metastasis models. The findings showed that depletion of USP36 exhibited smaller tumors, which notably inhibited tumor weight and volume (Figs. 6A and S4A). Conversely, ectopic expression of USP36 had the opposite effects. IHC revealed that USP36 depletion decreased Ki67 expression, while its overexpression led to an upregulation of Ki67 (Figs. 6B and S4B). Furthermore, in liver and lung metastasis models, lower luciferase intensity and fewer metastasis nodules were observed in the USP36-depleted group, whereas ectopic expression of USP36 exhibited an opposite phenomenon (Figs. 6C–F and S4C, D). Taken together, these findings demonstrate the involvement of USP36 in the promotion of CRC proliferation and metastasis in vivo.
Fig. 6: USP36 promotes CRC proliferation and metastasis in vivo.
A Representative photographs of subcutaneous xenograft tumors were obtained from nude mice. The tumor volumes were measured every 5 days and the tumor weights were analyzed. B HE and IHC staining of xenograft tumors from HCT 116 and LoVo cells. The expression of Ki67 was detected by IHC. C, D Representative photographs and analysis of luminescence intensity in metastasis models (n = 5 for each group). E, F Representative photographs and HE staining of metastatic tumors in the livers and lungs of mice. The number of metastases in livers or lungs was analyzed. All data are presented as the means ± SD of three independent experiments. ***p < 0.001.
USP36 positively correlates with RBM28 in CRC samplesTo further investigate the clinical relevance of USP36 and RBM28, we initially examined their protein abundance and correlation in human CRC samples through Western blot and immunohistochemistry (IHC). Firstly, Western blotting analysis revealed that both USP36 and RBM28 expression levels were significantly elevated in CRC compared to normal tissues (Fig. 7A), with a significant positive correlation observed between their protein levels (n = 24, Pearson R = 0.5165, p = 0.0098) (Fig. 7B). Subsequently, IHC staining was performed to validate their expression in tissue microarrays (TMAs) containing 80 paired tissues from CRC patients (Fig. 7C, E). As shown in Fig. 7D, F, the analysis of IHC scores demonstrated a significant increase of USP36 and RBM28 in CRC tissues. Furthermore, representative staining images implied a notable positive correlation between USP36 and RBM28, substantiated by Pearson’s correlation coefficient (R = 0.7689, p < 0.0001) (Fig. 7G, H). These results indicated that USP36 and RBM28 are upregulated in CRC and positively correlated with each other.
Fig. 7: USP36 positively correlates with RBM28 in CRC samples.
A Western blot analysis of cell lysates from 24 paired CRC samples using USP36 and RBM28 antibodies. B Correlation analysis of USP36 and RBM28 in CRC samples (n = 24). The chi-square test was applied to statistical analysis. Pearson R indicates the correlation coefficient. C, E The protein level of USP36 and RBM28 was detected by IHC in the TMA of 80 paired CRC patients’ samples (left panel). Representative images of high USP36 and RBM28 expression in CRC tissues (right panel). D, F The IHC score of USP36 and RBM28 in the TMA of 80 paired CRC patients’ samples. The comparison between cancer tissues and matched adjacent tissues was conducted using either the Wilcoxon unpaired test (left panel) or paired t-test (right panel). G Representative images of IHC staining of simultaneous high or low expression of both USP36 and RBM28 on the tissue of CRC patients. H USP36 IHC-scores (X-axis) positively correlate with RBM28 IHC-scores (Y-axis) in CRC samples (n = 80). The p value was calculated from a linear regression analysis. R is the correlation coefficient. I, J Kaplan–Meier plots of OS in the TMA of 80 paired CRC patients, divided into “high” and “low” expression groups based on the median IHC score of USP36 and RBM28. The p value was calculated from a log-rank test. HR hazard ratio. All data are presented as the means ± SD of three independent experiments. ***p < 0.001.
Based on these results, we further analyzed the relationship between the IHC score of the two proteins and clinicopathological characteristics in the TMA. This analysis revealed that USP36 was positively correlated with tumor stage (p = 0.0008), lymph node metastasis (p = 0.0066), vascular invasion (p = 0.0181), and distant metastasis (p = 0.0026) (Table S1). Likewise, RBM28 also showed positive correlations with tumor stage (p = 0.0136), lymph node metastasis (p = 0.0237), vascular invasion (p = 0.0181), and CEA levels (p = 0.0389) (Table S2). More importantly, both high levels of USP36 and RBM28 were positively correlated with tumor stage (p = 0.0020), lymph node metastasis (p = 0.0122), vascular invasion (p = 0.0210), distant metastasis (p = 0.0192), and CEA levels (p = 0.0380) (Table S3). In addition, Kaplan-Meier analysis of TMA showed shortened overall survival in patients with higher USP36 or RBM28 compared to those with low levels (Fig. 7I, J). Collectively, these data demonstrated that elevated levels of USP36 and RBM28 correlate with poor overall survival in CRC, highlighting their potential as pivotal biomarkers for diagnosis and prognosis for CRC.
RBM28/p53 axis mediates USP36-induced proliferation and metastasisTo evaluate whether USP36-induced biological functions were mediated by RBM28, we transfected stably depleting USP36 cells or control cells with RBM28 or vector. Meanwhile, lentivirus-mediated shRBM28 or control was transfected into cells with USP36 overexpression. In CCK8 and colony formation assays, elevated RBM28 enhanced cell proliferation and reversed the cell growth decreased by USP36 knockdown. Conversely, RBM28 depletion attenuated cell proliferation and restored the enhanced cell growth caused by USP36 overexpression (Fig. S5A–D). Meanwhile, flow cytometry apoptosis assays revealed that increased RBM28 resulted in a lower apoptotic rate in CRC cells and restored the increased apoptotic rate caused by USP36 silencing, and vice versa (Fig. S5E, F). In addition, upregulated RBM28 enhanced the metastatic ability of CRC cells and counteracted the weakened effects resulting from USP36 depletion, and vice versa (Fig. S6A–D). Furthermore, in vivo xenograft tumor assays revealed that RBM28 silencing exhibited smaller tumors and reversed the promoted tumor growth induced by USP36 expression, as observed in alterations in tumor volume, weight, and Ki67 levels (Fig. 8A, B). Similarly, in vivo metastasis assay revealed that RBM28 silencing led to a decrease in the number of liver and lung metastatic nodules induced by USP36 overexpression, indicating RBM28 reversed the pro-metastatic ability caused by USP36 overexpression (Fig. 8C–F). Taken together, these results suggested that USP36 promotes proliferation and metastasis in CRC through stabilizing RBM28.
Fig. 8: USP36 promotes the growth and metastasis of CRC cells in an RBM28-dependent manner.
A Representative photographs of subcutaneous xenograft tumors were obtained from nude mice. The tumor volumes were measured every 5 days and the tumor weights were analyzed. B HE and IHC staining of xenograft tumors. The expression of Ki67 was detected by IHC. C–F Representative photographs and HE staining of metastatic tumors in the livers and lungs of mice. The number of metastases in livers or lungs was analyzed. G A schematic model for the mechanisms of USP36 in CRC. USP36 deubiquitinates and stabilizes RBM28 at the K162 residue. The increased RBM28 then interacts with p53, inhibiting its transcriptional activity and leading to the inactivation of the p53 signaling pathway. All data are presented as the means ± SD of three independent experiments. ***p < 0.001.
Next, we evaluated whether USP36/RBM28 exerts its oncogenic effects through the p53 signaling pathway. Through CCK8 and colony formation assays, we found that Idasanutlin (a p53 activator) markedly suppressed cell proliferation and counteracted the increased cell growth driven by the overexpression of USP36 and RBM28 in LoVo cells (Fig. S7A, B). Similarly, flow cytometry apoptosis assays also indicated that Idasanutlin promoted apoptosis of LoVo cells and reversed the reduced apoptotic rate caused by USP36 and RBM28 overexpression (Fig. S7C). Furthermore, Idasanutlin notably inhibited the metastatic capability of LoVo cells and restored the pro-metastatic effects of USP36 and RBM28 overexpression (Fig. S7D, E). Collectively, these results demonstrated that USP36/RBM28 facilitates oncogenic processes through the p53 signaling pathway and highlighted the potential of Idasanutlin to target this oncogenic pathway in CRC cells.
Comments (0)