
Remember me


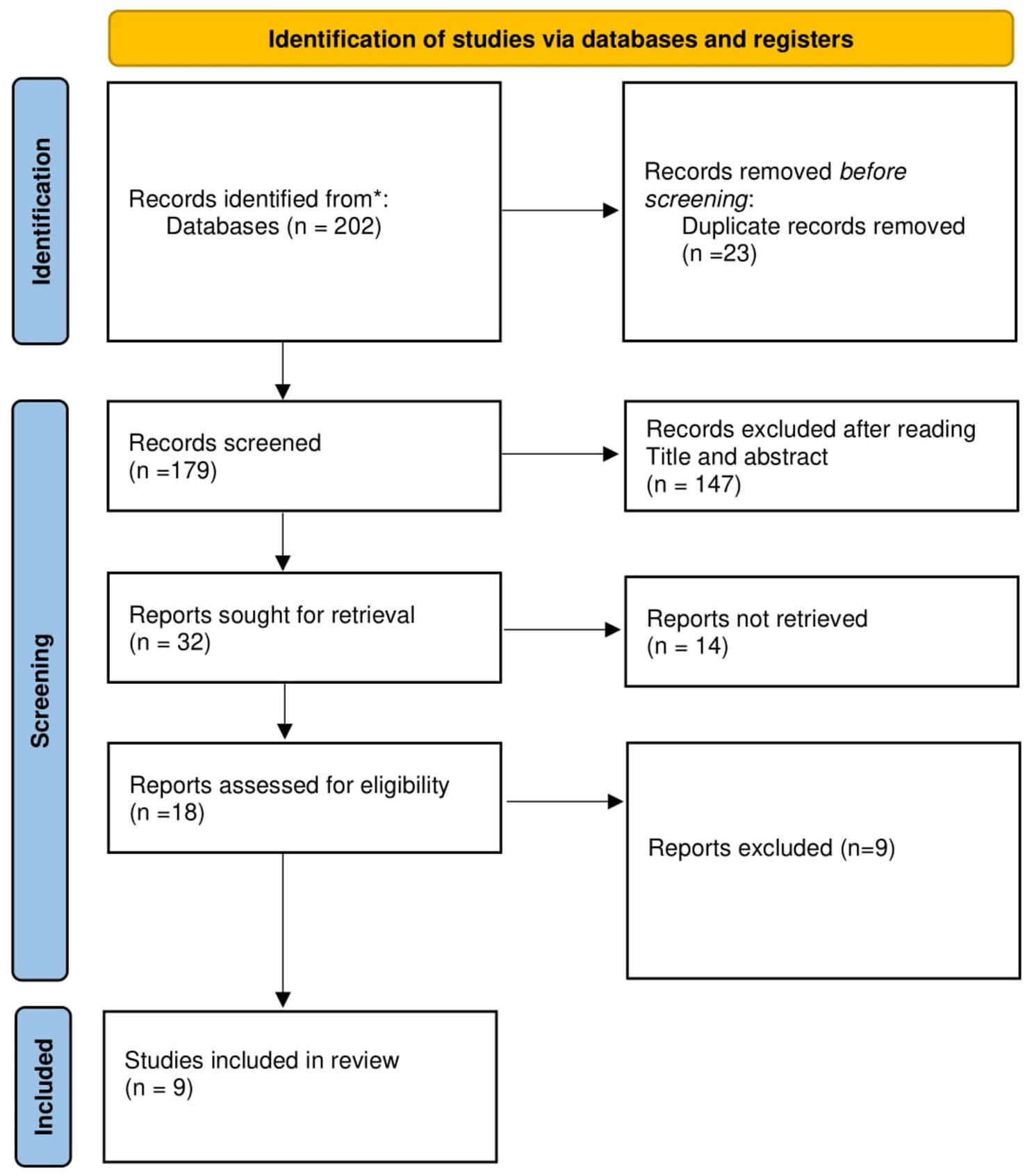

A total of 20 co-crystal structures of GSK-3β were downloaded from the PDB (Table 1). Taking into account the observed interactions between the individual ligand and its respective binding pocket, the structures were aligned to all co-crystal ligands, and features common to all such ligands were identified using PyMOL. Binding poses of each aligned ligand are shown in Fig. 2A. There were five common features among all the residues that interacted with the key residues as presented in Fig. 2B.
Table 1 The co-crystal structures used in the studyFig. 2
A The alignment of co-crystal ligands to extract common pharmacophoric features. B The common pharmacophoric features represented on template ligand (6LQ). Orange circles show aromatic rings, cyan spheres show hydrogen bond donors, and red sphere shows hydrogen bond acceptor
Pharmacophore Query Generation and ValidationA pharmacophore hypothesis was created with a co-crystal ligand (PDB ID: 6LQ) as a template. The crystal template was brought into the Maestro workspace, and shared pharmacophoric features were determined based on ligand–protein interactions. The query that was developed was validated using the AUC value, which effectively discriminated active ligands from decoys with an AUC score of 0.82. A set of 200,000 compounds in the VITAS-M Lab database were screened against this confirmed pharmacophore hypothesis. The analysis conducted at EF1% showed that pharmacophore-based screening is suitable for enriching active inhibitors for GSK-3β. About 50 out of 174 identified actives were retrieved in the top 2000 compounds from the virtual screening results. This gave us our calculated value for EF1% of − 28.73, indicating that the top 1% of ranked compounds had 28.73 times the number of active ligands than would be expected from random selection. Hence, this validated that our virtual screening approach successfully prioritized high-affinity ligands, thereby ascertaining the strength of the pharmacophore model for the discovery of potential ATP-competitive GSK-3β inhibitors. Compounds were designated as potential hits if they passed at least four critical pharmacophoric features. Screening outcomes were ordered by the Phase screen score, which integrated vector alignment, volume score, and RMSD site match. The Phase screen score was between − 1.0 and 2.0, and higher values were indicative of improved alignment. Using a cutoff score of 1.7, 174 compounds were chosen for further molecular docking experiments. These highest-scoring compounds were structurally compatible with the pharmacophore model to ensure a precise selection for additional binding affinity measurements. The refined pharmacophore hypothesis and screening outcome are shown in Fig. 3.
Fig. 3
A The area under curve of validated query differentiating active ligands from decoys with a value of 0.82. B The optimized pharmacophore hypothesis
Molecular DockingMolecular docking of the selected ligands against GSK-3β were conducted with Glide GScore and RMSD serving as parameters for the strength of the binding poses. Based upon the GScores, four compounds (VL-1, 2, 3, and 4) were chosen, and their molecular interactions were analyzed (Table 2 and Fig. 4; see also Supplementary Figure for superimposition image). VL-1 formed four hydrogen bonds with Val135, Asp181, Lys183, and Asp200, along with five pi-alkyl interactions and one pi-sulfur bond. VL-2 created three hydrogen bonds with Lys85, Asp133, and Val135, along with four alkyl bonds and one pi-sulfur bond. VL-3 established one hydrogen bond with Val135, and VL-4 formed two hydrogen bonds with Lys85 and Asp200. Additional exploration of potential binding modes is depicted in Fig. 5.
Table 2 The glide gscore and RMSD values of the selected docked compoundsFig. 4
The molecular interactions of hit compounds with the binding pocket of GSK-3β enzyme. Hydrogen bonds are shown with green color, Pi-Sulfur bonds with orange, hydrophobic interactions with pink color spheres
Fig. 5
The potential binding configurations of the active compounds within the GSK-3β binding pocket. The residues of the binding site are illustrated in soft green sticks, while the hit ligands are represented with sticks in various colors
ADMET PropertiesSubsequent to the docking analyses, the four compounds were subjected to assessment of their physicochemical and ADMET (absorption, distribution, metabolism, excretion, and toxicity) parameters. The physicochemical parameters were calculated using QikProp software (Ioakimidis et al. 2008). All compounds adhered to Lipinski’s Rule of Five (Pollastri 2010), indicating good drug-like potential. These included characteristics such as optimal molecular weights (≤ 500 Da), hydrogen bond donors (≤ 5), and acceptors (≤ 10). Further, the predicted octanol/water partition coefficient (QPlogPo/w) values were ≤ 5, indicating optimal lipophilicity. The permeability through Caco-2 cells (QPPCaco) was found to be moderate to high for the four drugs, while the human serum albumin binding (QPKhsa scores) was also in the acceptable range. The brain-to-blood partition coefficient (QPlogBB) values ranged from − 2.3 to − 0.53, while the hERG K+ channel blocking activity (QPloghERG values) was measured between -7.1 and -5.1.A full summary of the physicochemical and ADMET properties of the docked compounds is presented in Table 3.
Table 3 The physiochemical and ADMET properties of the docked hitsMDSAfter ADMET analysis, top two hits (VL-1 and 2) with best binding modes and ADMET properties, were selected for the evaluation of their stability with GSK-3β protein. The binding modes of selected hits were aligned on template co-crystal ligand and are depicted in Fig. 6.
Fig. 6
The binding configurations of the active compounds as they align with the co-crystal ligand. In panel (A), VL-1 is depicted with blue sticks, perfectly aligned alongside the red co-crystal ligand. In panel (B), VL-2 is showcased using red sticks
The RMSD analysis was conducted by examining the trajectories, ensuring the stability of the complexes in comparison to solid protein–ligand complexes. The results illustrated that both complexes equilibrated by the 10 ns time point (Fig. 7A). After this equilibration, RMSD for the VL-1 complex fluctuated marginally from 20 to 40 ns, and stabilized at around 1.5–2 Å at 70 ns. After this equilibration period, only minor fluctuations could be seen, and the RMSD increased to around 2.25 Å at 80 ns, after which it leveled off and was stable for the rest of the simulation time. The RMSD of the VL-2 complex also leveled off after equilibration and remained between 1.75 and 2 Å until 80 ns. The root mean square fluctuation (RMSF) values were obtained for evaluating the flexibility of residues within the protein upon its interaction with the ligands. Throughout most of the simulation, the RMSF values for the individual residues stayed below 2 Å except for the loop regions, as shown in Fig. 7B. This indicates that protein residues were stable and showed minimal fluctuation during the simulation, confirming the stability of the ligand–protein complexes.
Fig. 7
A The RMSD graphs illustrating the behavior of the protein and ligands throughout the 100 ns simulation period. B The variations in protein residues during the simulations, assessed through RMSF values. The blue graph represents the RMSF of the VL-1 complex, while the green graph depicts the VL-2 complex
Protein–Ligand ContactsThe following were identified as the most significant categories of interactions between the target protein and the ligands, as determined by MDS: hydrogen bonds, ionic bonds, and hydrophobic interactions. The hydrogen bonding side chains with the VL-1 complex included Ser66, Lys85, Val135, and Lys183 (Fig. 8A). In contrast, Lys85, Asp133, Val135, and Asp200 made hydrogen bonds with the VL-2 compound (Fig. 8B).
Fig. 8
The interaction of protein–ligand during MDS. A The protein–ligand contacts of VL-1 complex. B The protein–ligand contacts of VL-2 complex. The interacting residues are shown are shown as stacked bars
PCAPrincipal Component Analysis (PCA) was utilized to examine the dynamic behavior of proteins in both complexes, focusing on the collective motions observed in the MD trajectories. The variance proportion was plotted against the eigenvalues to illustrate the dynamic motions in hyperspace. Only the first three principal components (PCs) were considered, as they accounted for the majority of the fluctuations. For the VL-1 complex, PC1 exhibited the highest variation, accounting for 20.21%, while PC2 and PC3 showed variations of 12.84% and 7%, respectively (Fig. 9A). In the case of the VL-2 complex, PC1 displayed the largest variation at 19.14%, followed by PC2 and PC3 with variations of 10.75% and 7.47%, respectively (Fig. 9B). PCA analysis also revealed conformational changes across all clusters in the PC subspace, with the blue regions indicating the most significant movements, while the red regions corresponded to areas with less flexibility and movement.
Fig. 9
Principal component analysis of VL-1 and VL-2 complexes. A The PCA plot of VL-1 complex with overall flexibility of 40.05%. B The PCA plot of VL-2 complex with overall flexibility of 37.36% in three hyper spaces
Cross CorrelationProtein residue interaction analysis was conducted using cross correlation matrix, with cyan and magenta colors representing positively correlated and anticorrelated residues. Most of the residues were positive correlated, as can be observed from the diagonal lines which show the positive correlation of topologically local residues (Fig. 10). In summary, our analyses indicate a strong correlation of protein residues with respect to specific compound binding during the simulation process.
Fig. 10
The dynamic cross correlation matrix of the protein complexes to find the positive correlation in protein residues. A The DCCM of VL-1 complex. B The DCCM of VL-2 complex
MM/GBSAThe MM/GBSA method was used to calculate the total binding free energy (ΔG_total) of both protein–ligand complexes. The energy calculation considered van der Waals (ΔE_vdW), electrostatic (ΔE_ele), and solvation (ΔG_GB) contributions. For the complexes, the Van der Waals and and electrostatic components were similar; however GB contributions were greater for the VL-1 ligand compared to VL-2. The calculated ΔG_total values were − 37.55 ± 0.50 for VL-1 and − 28.85 ± 0.33 for VL-2, indicating a greater stability of the former complex (Table 4). Figure 11 displays the energy component contributions.
Table 4 The Binding free energies of the complexes calculated by implying MM/GBSA moduleFig. 11
The contribution of binding energy components in total binding free energy
Free Energy Landscape (FEL) AnalysisFEL analysis was performed for both VL-1 and VL-2 complexes for provide further validation of the stability of ligand–protein complexes. The FEL of VL-1 (Fig. 12A) confirmed the presence of a single deep minimum at x = − 0.56445, y = − 5.79882, z = 0.0, suggesting that the complex remained very stable during the entire simulation. The existence of only one deep basin points to the presence of little conformational fluctuations with regards to the binding stability of VL-1 within the GSK-3β active site. In the case of VL-2 (Fig. 12B), two minima were observed in the FEL; at x = 2.27375, y = − 2.22768, z = 0.931, and at x = 3.40904, y = − 2.22768, z = 0.931, indicating the presence of two distinct stable conformations. The first minimum seems to suggest a dominant stable state, while the presence of the second minimum indicates the existence of possible conformational flexibility, which may permit VL-2 to adapt its binding mode inside the active site. Thus, the overall evidence from the FEL analysis confirms that both VL-1 and VL-2 are thermodynamically stable, with VL-1 exhibiting a more rigid binding mode, while VL-2 exhibits some flexibility in its binding conformation. This, in turn, supports the viability of both compounds as GSK-3β inhibitors, while also suggesting that VL-1 may have an advantage in stability for therapeutic application.
Fig. 12
Free Energy Landscape (FEL) analysis of VL-1 (A) and VL-2 (B) complexes. Minima points indicate stable conformations, with VL-1 showing a single deep basin and VL-2 displaying two stable states
Comments (0)