Despite the clear and pressing needs faced by those with T1D, some nonetheless claim that insulin replacement and diabetes technologies are so good that little risk should be tolerated when developing new T1D therapies [32]. We believe the data show otherwise. It is therefore imperative that we continue to bring innovative therapies to address the significant and serious unmet needs of those living with T1D.
Insulin replacement can no longer be seen as the only therapeutic option for T1D. We must aim to modify the overall course of T1D, employing targeted therapeutic strategies for each stage of the disease (Table 1). Ideally, multiple therapeutic options and modalities would be available at each stage, allowing people with T1D more choice in the most appropriate care for them. Some progress has been made in bringing novel types of therapies to market, for example, teplizumab and donislecel.
Table 1 Treatment strategies beyond targeting glycemic control, therapeutic goals, and ideal treatment characteristics for each stage of type 1 diabetesTeplizumab is an anti-CD3 monoclonal antibody and is the first disease-modifying therapy approved for use in T1D. Teplizumab is indicated to delay the onset of stage 3 T1D in children and adults aged ≥ 8 years with stage 2 T1D [33]. Donislecel is an allogeneic pancreatic cellular therapy indicated for the treatment of adults with T1D who are unable to approach their target HbA1c because of repeated episodes of severe hypoglycemia despite intensive diabetes management and education [34]. Donislecel is the first approved cell therapy available in type 1 diabetes but must be used in conjunction with concomitant immunosuppression. These therapies represent important first steps at moving beyond the insulin-centric paradigm of the last hundred years; however, more is needed to fully meet the needs of people with T1D.
Bringing novel treatments and cures to fruition requires new solutions to long-existing challenges. Researchers and regulators must move beyond the insulin-centric paradigm of the previous 100 years, and the regulatory environment must protect public health without suppressing innovation. Insulin-based treatments cannot remain the status quo—the T1D community can and should do better. We must also foster a health system that allows breakthrough therapies to be available and accessible to all who need them.
Regulatory Opportunities to Advance T1D Cures
Clear and reasonable regulatory pathways are essential components of a healthy, viable, and inviting product development ecosystem. This has not always existed in T1D, evidenced by the recent history of several sponsors halting or significantly diminishing their T1D programs in response to perceptions of an unfavorable regulatory landscape, in some cases even after positive clinical trial results.
Other autoimmune diseases have well-established regulatory pathways to move beyond treating disease symptoms to modifying the underlying disease itself. In rheumatology, for example, more than a dozen Disease-Modifying Antirheumatic Drugs, or DMARDs, are available. People with rheumatoid arthritis no longer rely solely on painkillers, as was the case in the first half of the twentieth century. Solutions to regulatory challenges in T1D have been identified, and we must work together urgently to implement these solutions.
Assessing Benefits and Risks Through the Eyes of Those with T1D
Decisions to approve any new therapy fundamentally center on whether the benefits of a therapy justify the associated risks. Benefits must be clinically meaningful to people living with the disease, and risks must be tolerable in the short- and long-term when measured against the risks and burden of living with the disease. A product can only be approved when the totality of available evidence, along with the uncertainties in that evidence and risk management strategies, supports a favorable benefit-risk assessment [35].
The benefit/risk paradigms employed by regulators often fail to account for the lived experiences and outcomes of those living with T1D. A product’s risks and benefits cannot be weighed against the view that T1D is adequately managed with insulin and technology. This does not reflect the reality for most with T1D and prevents innovation toward new therapeutic regimens. The magnitude of benefits required to outweigh a product’s risks should be determined by people living with T1D and no one else.
Incorporating patient perspectives into regulatory decision-making, often called “patient-focused drug development,” helps facilitate patient enrollment, reduces patient burden in clinical trials, and informs the acceptability of tradeoffs between treatment benefits and risks, among other stated benefits [36]. For example, an investigational product may demonstrate equivalent HbA1c effects as standard-of-care therapy but decrease diabetes-related distress or fear of hypoglycemia. It would, therefore, benefit patients and improve the standard of care, assuming all else was equal. Unfortunately, there is currently an absence of validated measures accepted by regulators to inform approvals that capture patient perspectives to inform these decisions.
Providing robust patient preference data on risk tolerance also ensures important regulatory decisions align with the views of patients. A salient example is seen in cell replacement therapies, which often result in complete insulin independence or reliance on little insulin while providing more day-to-day predictability and protection from hypoglycemia. The only available form of this kind of therapy requires using long-term immunosuppression, which is associated with significant adverse events and included as part of the overall risk of cell replacement therapy. However, it is imperative that regulators do not overweigh the risk of immunosuppression in the context of the risks of living with T1D without adequately determining the preferences of those living with T1D.
Another example involves the selection of a clinical trial population. In most cases, the current regulatory perceptions of the risks associated with cell therapies for T1D only allow for participation by those with the most severe cases of T1D—with frequent severe hypoglycemia or hypoglycemia unawareness. However, given the daily lived experience of those in the T1D community, it is reasonable to expect many would welcome the opportunity to join a clinical trial that offered respite from the burdens of insulin, despite the possibility of trading old risks for those associated with novel therapies. Limiting clinical trials to select and small populations lengthens the development pathway, discourages additional innovation and investment, and slows the pace of cures becoming available.
Validation of New Endpoints
HbA1c and hypoglycemic events are the primary measures accepted by regulators to assess therapies targeting glycemic control. These outcomes are well suited for this purpose, but they are not ideal endpoints for trials of novel curative treatment approaches.
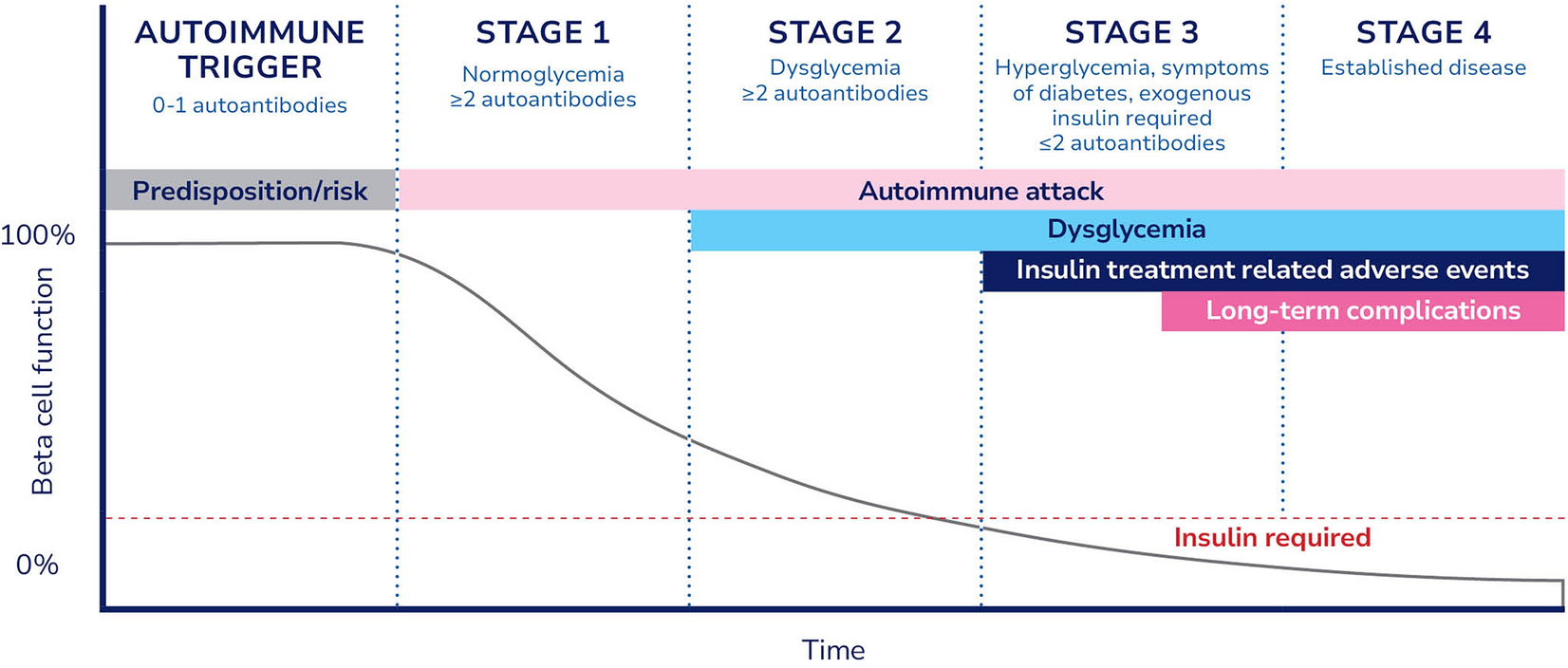
For example, disease-modifying therapies aim to preserve existing beta cell function, which is well established to improve T1D outcomes, including lowered hypoglycemia, improved HbA1c, and reduced long-term complications [37]. In trials of disease-modifying therapies, especially in the early period after insulin is required (i.e., new-onset stage 3 T1D), HbA1c and hypoglycemia are not practical or ideal endpoints. With the increasing utilization of diabetes technologies, the heterogeneity of stage 3 T1D, and the variability in insulin requirements across a population, trials using these endpoints must be large and/or lengthy, decreasing feasibility and strongly disincentivizing product development. The established surrogate endpoints are impractical and introduce unnecessary barriers for product developers.
The T1D research community and experts have widely accepted that measuring beta cell function and insulin production, typically via C-peptide levels, is a more appropriate trial endpoint [38, 39]. C-peptide is a direct byproduct of insulin production, produced and secreted in equimolar concentrations to insulin, and is not a component of exogenous insulin formulations. Changes in C-peptide are also easier to assess in the timeframe of a clinical trial. Evidence supporting the validation of C-peptide continues to be published [37], including a recent meta-analysis of participant-level data from 21 randomized control trials [39].
However, the US Food and Drug Administration (FDA) has stated that C-peptide is only acceptable as a “reasonably likely surrogate endpoint,” capable of supporting accelerated approval. This requires post-marketing phase 4 studies and limits the viability of some drug programs [40]. Similarly, the European Medicines Agency (EMA) considers C-peptide an unvalidated surrogate endpoint that cannot support marketing authorization. As such, trials must continue to be designed with endpoints that complicate and lengthen development.
The advent of CGMs has also allowed for more direct and timely measurement of glucose levels than HbA1c can provide. Data collected by CGMs provide significant information regarding glycemic status, standardized CGM-derived metrics are used by patients and clinicians to guide day-to-day management, and CGM data are acknowledged and accepted by patients and healthcare providers as informative for managing T1D [41]. CGM-derived metrics could provide valuable and efficient means to assess the effects of novel therapies, and international consensus has been published on their role in clinical trials [42]. However, most CGM metrics, including those recommended by international consensus, are not currently accepted as primary endpoints by most regulators.
Additionally, patient-reported outcomes (PROs), clinically important aspects of care reported directly by the patient without interpretation or amendment from any others [43], can be used as primary or secondary outcomes in clinical trials to support product approvals if appropriately validated. Of the over 200 T1D PRO measures reported in scientific literature, only the Insulin Dosing Systems Perceptions, Ideas, Reflections, and Expectations (INSPIRE) tool [44] is accepted by regulators as part of medical device development. None of the existing PROs have been validated for use in drug or biologic trials.
Access to Expedited Programs
Finally, the pace of progress is directly related to the number of companies researching T1D and the magnitude of their commitment to the disease. To meet the pressing needs of those with T1D, regulatory pathways intended for products that treat serious and life-threatening diseases must be clearly available. Clearly defined T1D drug development tracks will help entice sponsors and attract increased investment.
The examples discussed here provide a glimpse into a few approaches to improving drug development in T1D, how we measure benefit, and how we benchmark risk. Many other opportunities have been identified that can further streamline the development pipeline, including better screening and early detection, trial enrichment strategies, advancing the role of relevant biomarkers, and increasing clarity on regulatory thinking on these and other topics.

Comments (0)