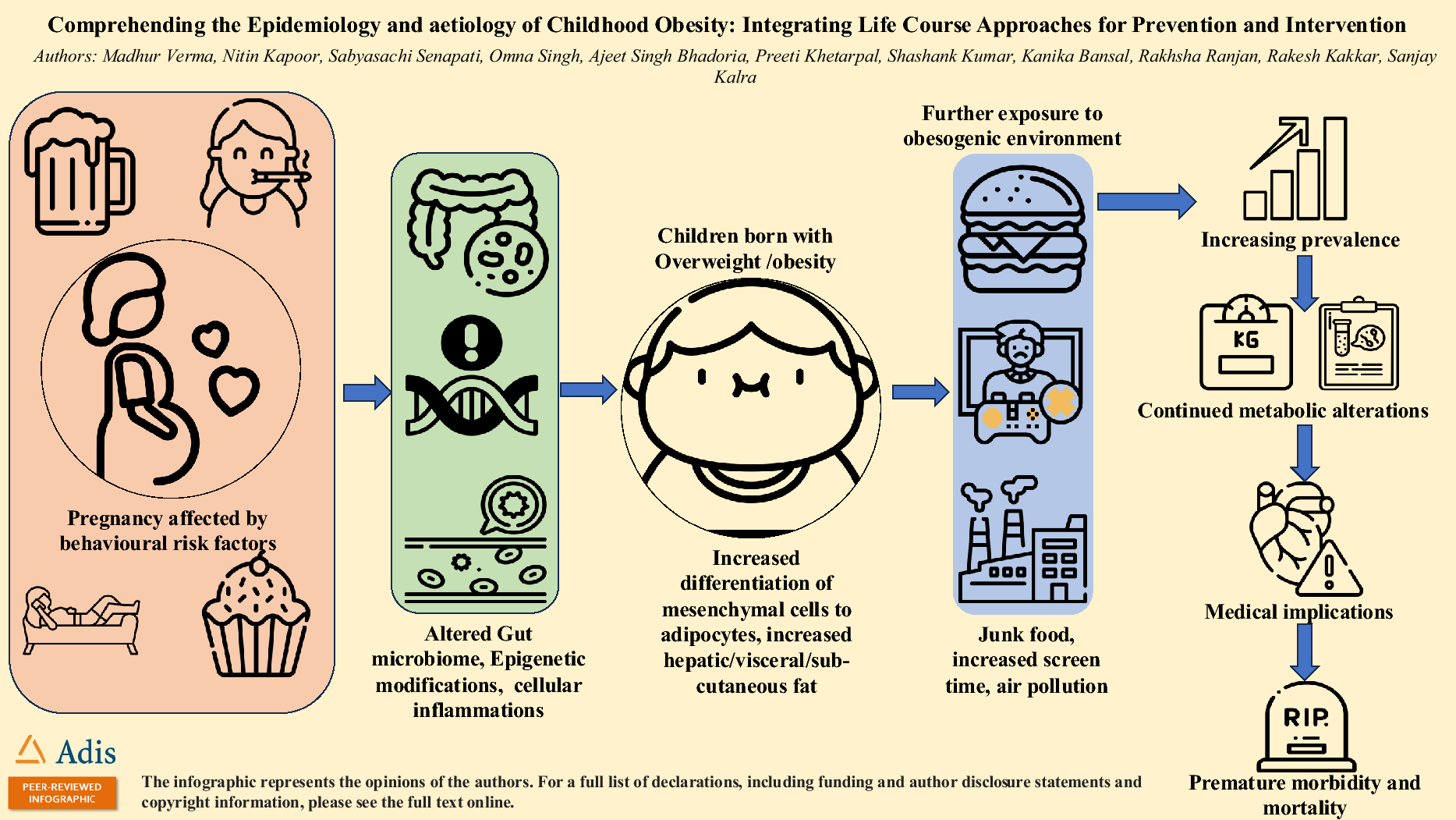
Study Design
This was a phase 1, randomized, double-blind, placebo-controlled, two-part study in healthy participants (November 2020 to August 2021) [Figure S1].
Part 1: a single-ascending dose (SAD) study, and
Part 2: a multiple-ascending dose (MAD) study with a drug–drug interaction analysis.
All participants had screening visits to confirm their eligibility. Screening occurred up to four weeks before enrolment and before randomization to the SAD or MAD study. The study was conducted in compliance with the principles of the Declaration of Helsinki, the Council for International Organizations of Medical Sciences International Ethical Guidelines, and the International Council for Harmonization Good Clinical Practice guidelines. The protocol was reviewed and approved by the Midlands Independent Review Board and the study was registered on ClinicalTrials.gov (NCT04559568). All participants provided written informed consent before participation.
Single-Ascending Dose Study
Six participants were randomly assigned to single-ascending oral doses of LY3522348 and two participants to placebo in five different cohorts (cohort 1: 5 mg; cohort 2: 15 mg; cohort 3: 50 mg; cohort 4: 150 mg; cohort 5: 380 mg; Figure S1). In the SAD study, a sentinel dosing approach was utilized for cohorts 4 and 5. Dose escalation to the next cohort was initiated following a satisfactory review of the safety, tolerability, and PK data from all previous cohorts. PK, PD, and safety assessments were performed at predefined times, including discharge from the investigative site on day 7 and a follow-up visit on day 14 (± 1).
Multiple-Ascending Dose Study
The MAD study was initiated after assessing the safety and tolerability of LY3522348 doses in cohort 4 and PK and PD data in cohort 3 in the SAD study. In the MAD study, six participants were randomly assigned to once daily (QD) oral doses of LY3522348 and two participants to placebo for 14 or 15 days in three different cohorts (cohort 1: 50 mg; cohort 2: 120 mg; cohort 3: 290 mg; Figure S1). PK, PD, and safety assessments were performed at predefined times till discharge from the investigative site on day 20 for cohort 1 and 2 or day 21 for cohort 3 and during a follow-up visit on day 28 (± 2). In addition, as an extrapolatory study to assess the potential impact of LY3522348 treatment on CYP3A metabolism, participants in cohort 3 received a single oral dose of 200 μg midazolam, a CYP3A substrate, on day – 1 and within 15 min of LY3522348 or placebo dosing on day 15. Figure S1 provides details on the study duration, doses, dosing strategy, participant admission, and treatment.
Study Population
Healthy men and women aged 18–65 years who were not of childbearing potential, had a body mass index between ≥ 18.5 kg/m2 and ≤ 40 kg/m2, had a stable weight for 1 month prior to screening and enrollment, and had safety laboratory test results within the normal reference range for the population were considered for inclusion. Participants were excluded if they had an abnormality in the 12-lead electrocardiograms (ECG) at screening that may confound ECG analysis, had a blood pressure of > 160/90 mmHg and pulse rate < 50 or > 100 bpm, had a history of fructosuria, or used strong inducers or inhibitors of CYP3A or over-the-counter or prescription medication including herbal medications (St. John’s wort and vitamin/mineral supplements) within 14 days prior to dosing/start date (Figure S1).
AnalysesDemographics and Participant Disposition
Participant disposition and demographic variables such as age, sex, race, ethnicity, body weight, height, and body mass index were reported.
Safety Analyses
Safety was evaluated throughout the study period by monitoring for adverse events (AEs), physical examination, clinical safety laboratory assessments, vital sign measurements, and 12-lead ECG.
Pharmacokinetic Analysis
LY3522348 plasma and urine concentrations were determined using liquid chromatography-tandem mass spectrometry (LC–MS) at PPD Laboratories, WI, USA, and Q2 Solutions, Ithaca, NY, USA, respectively.
In the SAD cohorts, drug plasma concentrations were quantified from pre-dose to 144 h post-dose. In the MAD cohorts, drug plasma concentrations were quantified following the day 1 dose (pre-dose to 24-h post-dose), prior to the day 7 dose, and following the last dose (pre-dose to 144 h post-dose). Plasma PK parameters calculated included area under the concentration–time curve (AUC) from time zero to infinity (AUC(0–inf)), AUC over one dosing interval (AUCτ), maximum observed drug concentration (Cmax), time of Cmax (tmax), apparent volume of distribution during the terminal phase after extravascular administration (Vz/F), half-life associated with the terminal rate constant in noncompartmental analysis (t1/2), apparent total body clearance calculated after extravascular administration (CL/F) and accumulation ratio based on AUCτ and Cmax. The dose proportionality of LY3522348 PK was assessed separately for the SAD and MAD study, using day 14 PK data in the MAD assessment.
Plasma concentrations of midazolam and its metabolite, 1ʹ-hydroxymidazolam, were assayed on days − 1 (pre-dose to 24-h post-dose) and day 15 (pre-dose to 24-h post-dose) using validated LC–MS methods at Q2 Solutions, Ithaca, NY, USA. The impact of LY3522348 treatment on AUC(0–inf), Cmax, and tmax was evaluated.
Pharmacodynamic Analysis
A fructose tolerance test (FTT) was performed on day 1 in the SAD study and days 1 and 14 in the MAD study in which a fructose beverage was served with a low fructose meal (20 min, 6 h, 12 h post LY3522348 or placebo dose). Fructose plasma concentrations were measured following fructose beverage administration and these concentrations were used to calculate a fructose AUC(0–24).
Statistical Analysis
All analyses were performed using SAS® Version 9.4. For continuous data, mean, standard deviation (SD), median, minimum, maximum, and number of participants were reported. For log-normal data, such as AUC and Cmax, geometric mean, and geometric coefficient of variation were reported. For categorical data, frequency and percentages were presented.
PK parameter estimates were calculated using standard noncompartmental analysis methods using Phoenix WinNonlin Version 8.1.1. To estimate ratios of dose-normalized geometric means and corresponding 90% CIs, log-transformed LY3522348 PK parameters were evaluated using a power model (log-dose acted as an explanatory variable). The estimated ratio of dose-normalized geometric means of PK parameters between the highest and lowest doses was used to assess dose-proportionality. Renal clearance of LY3522348 was calculated as the amount excreted over 24 h (Ae[0–24]) divided by the plasma AUC over the same 24 h in the SAD study only.
In the SAD study, log-transformed FTT and biomarker parameters (low- and high-density lipoproteins, cholesterol, and triglycerides) were analyzed in an ANOVA model, with treatment as a fixed effect. In the MAD study, log-transformed FTT parameters were analyzed using a mixed model with repeated measures, with treatment, day, and treatment-by-day interaction as fixed effects and participants included as a random effect. The difference in the least square (LS) means between LY3522348 and placebo, along with the 90% CIs, were back-transformed to produce the ratio of geometric means and the CIs comparing LY3522348 to placebo by day. In the analysis of the effect of LY3522348 on midazolam and 1ʹ-hydroxymidazolam, PK parameters were assessed using an ANOVA model (the day was included as a fixed effect). The ratio of geometric means and corresponding CIs, along with the p value were also reported. Wilcoxon signed-rank test was used to analyze the tmax nonparametrically. The median of the differences comparing day 15 to day − 1 and the corresponding 90% CIs were presented alongside the p value. p values were not adjusted for multiplicity considering the phase and exploratory nature of the study. Missing data was not presented in the final analysis.

Comments (0)