The genetic era has led to an increasing number of relatives at risk of DCM coming to clinical attention. To the best of our knowledge, this study is the first to evaluate the yield of clinical DCM diagnosis and the possible optimisation of family screening for DCM in the Netherlands.
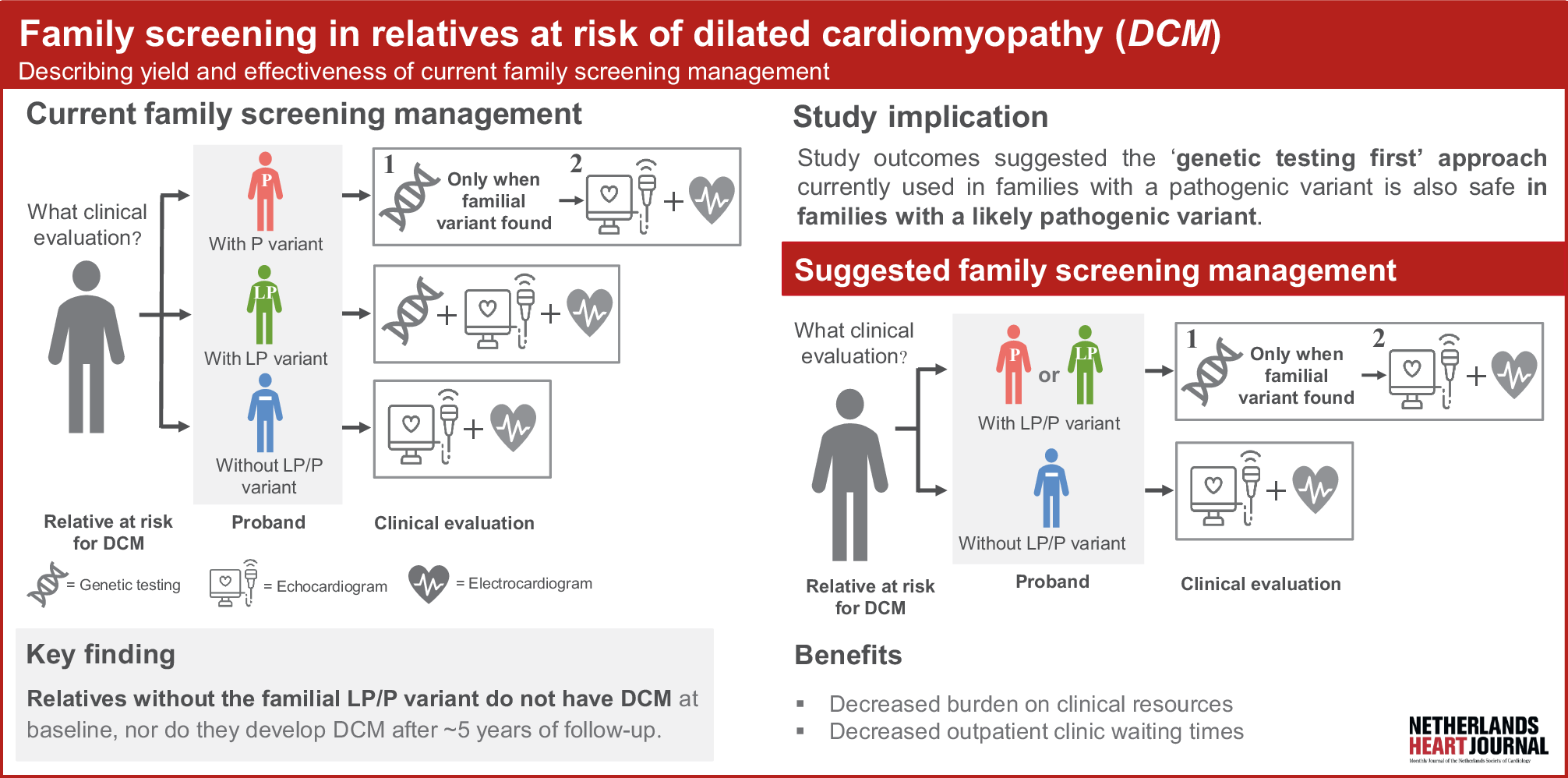
This study has several interesting results. First, the yield of screening at both baseline and after 5 years of follow-up was ~10%. Additionally, an age-related penetrance of DCM was observed. The yield was notably low in those < 20 years of age, with only 1 relative diagnosed with DCM who also had a proband diagnosed at infancy. This is in line with the results of a previous study [10] and implies that screening of relatives may be initiated at > 20 years of age unless the proband in the family was diagnosed before or during adolescence or an LP/P variant with known paediatric DCM onset is identified in the family [11].
Second, relatives without the LP/P familial variant did not have DCM at baseline, nor progression towards DCM during follow-up. These results are in line with a recent Danish study [12] and add to the body of evidence that relatives from LP families should first be offered genetic testing and can safely be discharged from cardiac screening when genetic testing proves to be negative (unless they are symptomatic). This also suggests that the 2023 ESC Guidelines for the management of cardiomyopathies may be safely implemented in the Netherlands.
Third, the baseline yield of DCM diagnosis in relatives of gene-elusive probands was unexpectedly high and comparable to those carrying an LP/P variant. As some of our cohort participants were relatives who received their genetic testing results more than a decade ago, this likely included probands who did not undergo genetic testing with the comprehensive panels and techniques we are currently offering. It is well known that updating genetic testing in the proband is beneficial in identifying the familial genetic variant [13, 14]. We therefore deem it likely that repeated genetic testing (which was not available for this study), would have identified an LP/P variant in a proportion of these families. This again reinforces the importance of updating genetic testing in DCM evaluation and shows that gene-elusive families may not be considered as gene-elusive in the future.
Last, our study shows that adherence to family screening ensures the diagnosis of DCM in all relatives prior to the occurrence of MACE, which is in line with previous reports (see Table S3 in Electronic Supplementary Material [10, 12, 15,16,17,18,19]) and family screening recommendations for other cardiomyopathies [3, 20,21,22,23,24,25]. Although this is reassuring, there is an increasing body of evidence suggesting that the clinical course of DCM is gene-specific, suggesting that gene-specific family screening algorithms may be beneficial [11, 26,27,28,29,30].
Study limitations and future perspectives
In this study population, we cannot exclude selection bias. For example, only 8 relatives carried the phospholamban (PLN) p.Arg14del variant, whereas a higher incidence was expected considering it is a Dutch founder variant. This could be explained by direct referral of PLN relatives to the University Medical Centre Utrecht rather than the Bergman Clinics because of its arrhythmogenic nature [27].
Next, the family size of relatives in our study ranged from 1 to 11 subjects. These large families could have skewed our results. Additionally, the study population consisted of only 14 relatives younger than 20 years since the Bergman Clinics did not routinely perform screening in paediatric subjects. Furthermore, we cannot rule out that relatives who were older and/or symptomatic were more likely to present for family screening, thereby leading to selection bias.
Moreover, the number of relatives from gene-elusive probands in our study was low, and the yield of screening in these individuals should be interpreted with caution. Last, it would have been of interest to investigate the gene-specific yield of family screening, as it is increasingly recognised that the clinical disease course is gene-specific. It will require large multicentre efforts to collect enough variant carriers per gene for meaningful analyses.






Comments (0)