Mouse models
All animal procedures and experiments were performed in accordance with national and European legislation, following approval by the 2nd Local Institutional Animal Care and Use Committee in Krakow (approval numbers 298/2017 and 61/2023). SLPI KO and WT mice were generously donated by Dr. Sharon M. Wahl [17, 18]. C57BL/6 mice were obtained from The Jackson Laboratory and housed under pathogen-free conditions in the animal facility at the Faculty of Biochemistry, Biophysics, and Biotechnology of the Jagiellonian University in Krakow. Littermate 10–12 weeks-old males were used for all experiments. A psoriasis model was induced using imiquimod (IMQ) [19]. Mice were treated twice daily for up to 6 days with 5 mg of Aldara (5% imiquimod cream) (Meda AB) applied to both sides of the depilated ear. For intravital imaging, mice were treated with IMQ for 2 days. Blood samples were collected into tubes containing 10 mM EDTA (Sarstedt). Bone marrow cells were isolated from femurs and tibias by centrifugation at 10,000 × g [20]. Ear biopsies were cut into small pieces and incubated with a 2.5 mg/mL Collagenase D (Roche Diagnostics) solution at 37 °C for 50 min. Single-cell suspensions from the ears were prepared by mashing the tissue through 40-µm cell strainers. Red blood cells were eliminated using RBC Lysis Buffer (eBioscience). The remaining cells were resuspended in RPMI 1640 medium (Biowest) supplemented with 2% heat-inactivated FBS (Gibco) and immediately stained for flow cytometry analysis.
Human samples
All human studies were performed in accordance with guidelines established by the Jagiellonian University Institutional Bioethics Committee under approved protocols (#87/B/2014; 1072.6120.30.2020) and adhered to the Declaration of Helsinki. Human blood was collected from healthy individuals who were fully informed and had consented.
Flow cytometry
Single-cell suspensions were stained for viability assessment (Zombie Aqua Fixable Viability Kit; BioLegend) and then unspecific antibody-binding sites were blocked with anti-CD16/CD32 antibodies (Fc block; eBioscience) followed by staining with directly conjugated antibodies: CD45.2-APC/Cy7 (BioLegend), CD11b-eFluor450 (eBioscience), Ly6G-APC (BioLegend) or c-Kit-PE/Cy7 (BioLegend), Lin-AF700 (BioLegend), c-Kit-APC/eF780 (eBioscience), CD34-FITC (eBioscience), Ly6G-BV711 (BD Biosciences). Data were acquired on a BD LSRII (BD Biosciences) and were analyzed using FCS Express (De Novo Software).
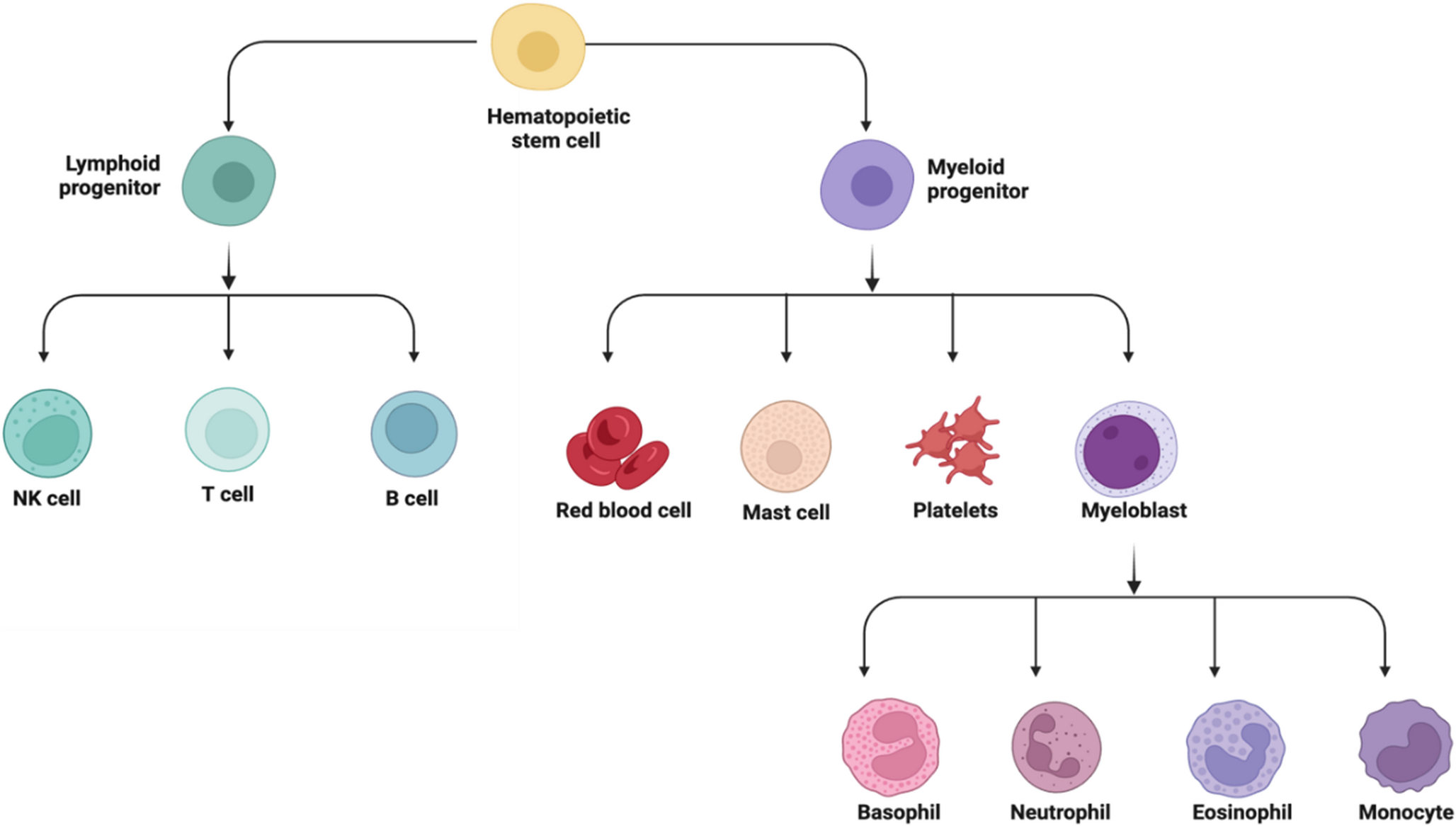
Singlets were selected on the basis of FCS-A vs. FCS-H. Dead cells were routinely excluded from the analysis. The frequencies of specific cell types were calculated as the percentage of CD45+ cells. Neutrophils were defined as live, CD45+, CD11b+ and Ly6G+ cells. Neutrophil progenitors were defined according to the gating strategy described previously [21]. The lineage antibody cocktail included anti-CD3 (clone 17A2), anti-Ly-6G/Ly-6 C (clone RB6-8C5), anti-CD11b (clone M1/70), anti-B220 (clone RA3-6B2) and anti-TER-119 (clone Ter-119) antibodies (BioLegend). c-Kit and CD34 were used to exclude CD34 – c-Kit+ stem/progenitor cells and FSC/SSC gating excluded eosinophils (SSChigh). The differential expression levels of c-kit and Ly6G were used to define the developmental stages of neutrophils. “Fluorescence minus one” (FMO) controls were routinely used to set the thresholds for positive/negative events.
Adoptive transfer
Bone marrow neutrophils were isolated by density gradient centrifugation. Diluted cell samples were layered on top of density gradient separation solutions (Pancoll human for Granulocytes 1.119 g/ml, Pancoll human 1.077 g/ml, and blood in a volume ratio of 1:1:2) (PAN Biotech). After centrifugation and washing, the remaining red blood cells in the granulocyte fraction were lysed using pyrogen-free water. The cells were then resuspended in RPMI 1640 medium (Biowest) supplemented with 10% heat-inactivated FBS (Gibco). The purity of the isolated cells was examined by flow cytometry based on CD45, CD11b, and Ly6G immunoreactivity, resulting in neutrophils with 64–80% purity.
The isolated neutrophils were labeled with CellTracker dyes (final concentration of 1 µM) for 15 min. at 37 °C and then washed twice with PBS (PAN Biotech) supplemented with bovine serum albumin (Sigma-Aldrich). CellTracker Red (CMTPX, Invitrogen) and CellTracker Green (CMFDA, Invitrogen) were used to differentially label neutrophils from WT and SLPI KO mice, respectively. Labeled neutrophils were resuspended in PBS (PAN Biotech) at a density of 1 × 108/ml, and 100 µl of cell suspension was injected intravenously (i.v.) via the jugular vein cannula 30 min. before intravital microscopy imaging. Each time, 1 × 107 neutrophils (mixed WT and SLPI-deficient neutrophils at a 1:1 ratio) were transferred per mouse.
Preparation of the mouse ear skin for intravital microscopy (IVM)
Mice were anesthetized with a mixture of ketamine hydrochloride (200 mg/kg; Biowet Puławy) and xylazine hydrochloride (10 mg/kg; Dechra) administered intraperitoneally. After anesthesia, cannulation of the right jugular vein was performed to maintain anesthesia and for the administration of antibodies or cells [22]. Preparation of the ear skin for intravital imaging was performed immediately after the injection of antibodies. Briefly, the mouse was moved to a custom-made Plexiglas board designed for microscopic imaging. The mouse was placed on its back, and the ear to be imaged was positioned on a stack of eight microscope slides (76 × 26 mm, 1 mm thick) taped together with surgical tape, with the ventral side up. Using a diamond glass-cutting knife, a coverslip was cut to the desired size and placed on the ventral side of the ear, with saline immediately applied underneath the coverslip.
Spinning disk confocal IVM
The mouse ear was visualized using a ZEISS Axio Imager.M2 upright microscope equipped with a metal halide light source (AMH-200-F6S; Andor, Oxford Instruments), a motorized 6-position excitation filter wheel, and a laser-free confocal spinning-disk device (DSD2; Andor, Oxford Instruments). The microscope utilized ZEISS EC Plan-NEOFLUAR 10×/0.3 and/or ZEISS EC Plan-NEOFLUAR 20×/0.5 air objectives. Four excitation filters were used: DAPI (390/40 nm), GFP (482/18 nm), RFP (561/14 nm), and Cy5 (640/14 nm), each visualized with the appropriate emission filters: DAPI (452/45 nm, exposure time 500 ms), GFP (525/45 nm, exposure time 500 ms), RFP (609/54 nm, exposure time 500 ms), and Cy5 (676/29 nm, exposure time 250 ms). Fluorescence detection was performed using a 5.5-megapixel sCMOS camera (Zyla 5.5; Andor, Oxford Instruments).
The iQ 3.6.1 acquisition software (Andor, Oxford Instruments) was used to control the microscope. A series of optical cross-sections (z-stacks) through the mouse ear skin was performed with a fully motorized microscope stage (Scan 130 × 85, ball screw pitch 2 mm; Märzhäuser Wetzlar). The z-step was set to 1 μm, and approximately 100 z-planes were captured, either in a single run or as a time-lapse acquisition over 10 min., with intervals of 750 ms, resulting in a total of 800 frames.
Imaging of neutrophils and blood flow with IVM
Imaging of neutrophils and platelets (to indirectly visualize blood flow) in the ear skin tissue and blood vessels was performed by staining the cells with appropriate fluorochrome-conjugated monoclonal antibodies. Neutrophils were visualized by injecting Ly6G-AF647 antibodies (1 µg per mouse; BioLegend) and Ly6G-eF450 antibodies (1 µg per mouse; eBioscience). Platelets were visualized by injecting CD49b-PE antibodies (0.6 µg per mouse; BioLegend). All antibodies were injected iv via the jugular vein cannula 30 min. before imaging, except for Ly6G-AF647 antibodies, which were injected 24 h before imaging (on the second day of IMQ treatment) via the tail vein. In experiments involving adoptive transfer, antibodies (Ly6G-AF647 and CD49b-PE) were injected 30 min. before imaging, just before the injection of CellTracker-stained neutrophils. Each mouse was imaged for up to 4 h, with separate movies/images acquired during this time.
The number of neutrophils in WT and SLPI KO mice was quantified both in circulation and outside of blood vessels nearby. Cells were counted in 5 different frames of 10-min. movies acquired with IVM, with each frame separated by 2 min. of movie. At least 2 different fields of view per mouse were analyzed from n = 3 mice per group.
The neutrophil migration rate through the blood vessel in the psoriatic ear skin was determined throughout the course of the acquired movies. In each 10-min movie, a line of approximately 50 μm was drawn inside the blood vessel (using iQ software, Andor, Oxford Instruments). Neutrophils that had slowed down were observed, and the number of frames needed for each cell to pass the given distance was counted and later converted into real time (seconds). One 10-min movie was analyzed for each mice from n = 3 mice per group.
Establishing neutrophil migration pattern in the psoriatic ear skin
The migration pattern and cell behavior in psoriatic ear skin tissue and blood vessels were analyzed using recorded movies of WT and SLPI KO mice. Each in vivo movie was opened in ImageJ (NIH) with the MTrackJ plug-in selected. For each movie, 20 cells were randomly chosen and tracked through the series of frames, marking their movement until they were no longer visible in the field of view (see supplementary Video). Each cell’s migration path was recorded in a different color, and the distance covered (in arbitrary units) was calculated automatically.
3D and 4D reconstruction of cross sections to analyze localization and movement of neutrophils
The number of neutrophils at different ear skin levels above the blood vessel of WT and SLPI KO mice was establish on a side view of a 3D reconstruction (IMARIS software, Bitplane, Oxford Instruments) of a series of optical cross-sections (z-stacks). A series of z-stacks through the ear skin was performed with a z-step of 1 μm approximately through 100 z-planes. Then, the ear structure was reconstructed with IMARIS software with red and violet color channels, for platelets indicating blood flow and neutrophils present in tissue for at least 24 h, respectively. Images were saved in. jpeg format and analyzed in ImageJ (NIH). An arbitrary mask, consisting of 3 frames each of approximately 17 μm length, was applied on images, and the number of neutrophils on three different levels above the blood vessel was establish.
Neutrophil migration pattern (4D) inside the ear skin was established by a time-lapse optical cross section acquisition (z-stack movies) through the mouse ear skin. The time-lapse series of optical cross sections was performed one after another for 10 min. (iQ software). Then, a 3D reconstruction in time (4D) was performed in IMARIS software (Bitplane, Oxford Instruments) and the z-stack movies were saved as. avi movie files and analyzed in ImageJ (NIH). In the latter software, 12 randomly chosen cells were followed through the time course of an entire movie and were tracked with the MTrackJ plug-in for ImageJ. A migration path was recorded for each cell and the distance (in arbitrary units) was calculated automatically. A minimum of 3 individual movies were analyzed per mouse for n = 3 mouse per group.
Human neutrophils isolation and migration analysis
Human blood was collected into sodium citrate and was subjected to isolation within 1 h of draw. Neutrophils were isolated by a density gradient 1.077 g/ml centrifugation, using Pancoll (PAN Biotech) and were recovered from the corresponding erythrocyte fraction of the Pancoll gradient, as described previously [7].
To study migratory behavior, neutrophils were seeded onto microscopy plates (µ-Plate 96 well black uncoated, Ibidi) and incubated for 15 min in RPMI1640 medium supplemented with 10% FBS, or with 10% FBS and 20 µg/ml recombinant SLPI [23]. Cells were then imaged for 30min. at room temperature using time-lapse video microscopy with a motorized stage (Olympus) at a frame rate of one frame every 16s.
The generated files were analyzed, identifying migrating cells as those with elongated shapes, adherent to the surface, that displayed any movement during the observation period. Cells were counted manually and expressed as a percentage of the total cells within the fields of view (FOV).
Preparation of human neutrophil extracts
Neutrophils isolated from the blood of healthy donors were plated on culture plates in RPMI1640 medium and incubated at 37 °C in a 5% CO2 atmosphere for 40 min. Recombinant SLPI [23] was added to the medium at a final concentration of 20 µg/ml, and the cells were incubated for an additional 30 min. under the same conditions. The cells were washed with PBS and centrifuged at 650 × g for 5 min. at 22 °C. The cell pellet was resuspended in relaxation buffer (10 mM PIPES, pH 7.3; 30 mM NaCl; 3.5 mM MgCl₂; 0.5 mM EGTA; 0.5 mM EDTA; and a freshly added protease inhibitor cocktail). Cells were then disrupted by cavitation in compressed nitrogen at 500 psi, centrifuged at 650 × g for 10 min. at 4 °C, and the pelleted lysates were resuspended in the relaxation buffer. Protein concentration in the samples was determined using the Bradford method. Lysates containing equal amounts of total protein were subjected to Western Blot analysis.
Recombinant SLPI was detected using primary anti-mouse His tag antibodies (Abcam) and secondary anti-mouse HRP antibodies (Abcam). A loading control was performed by detecting β-actin using primary anti-mouse β-actin antibodies (Abcam) and secondary anti-mouse HRP antibodies.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 9 (GraphPad Software). Data are shown as mean ± standard deviation (SD) or standard error of the mean (SEM). The specific tests performed and number of samples per group are described in the figure legends.









Comments (0)