
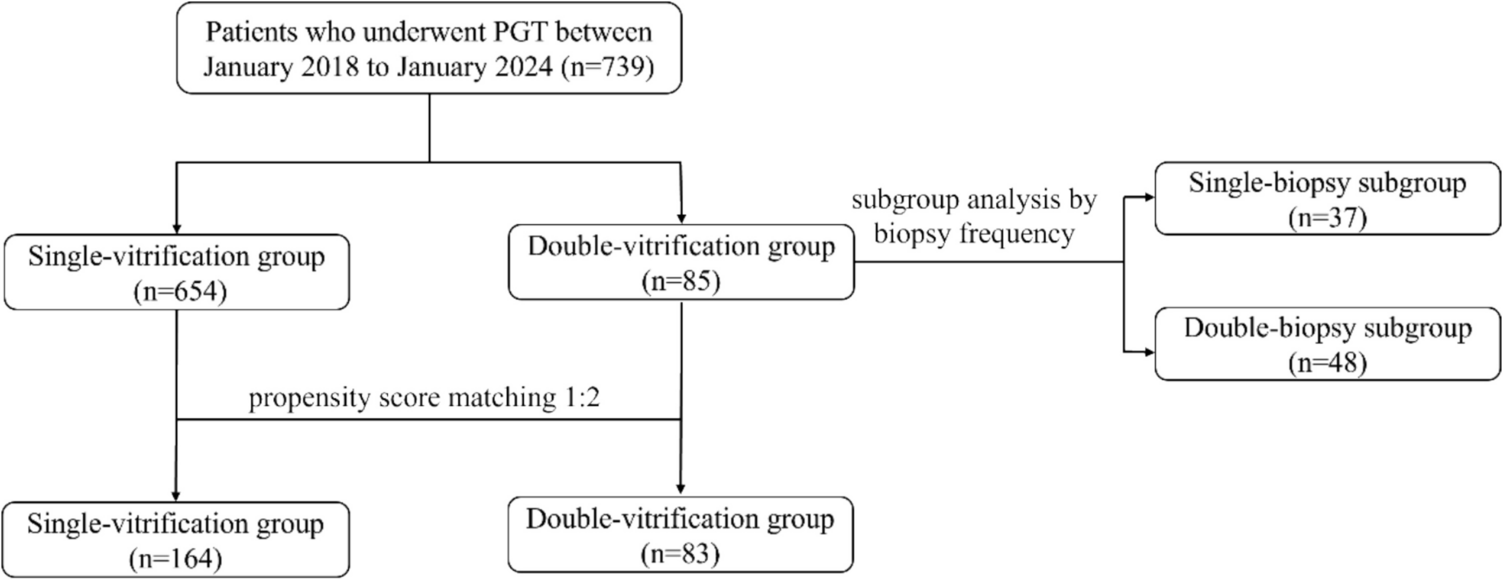
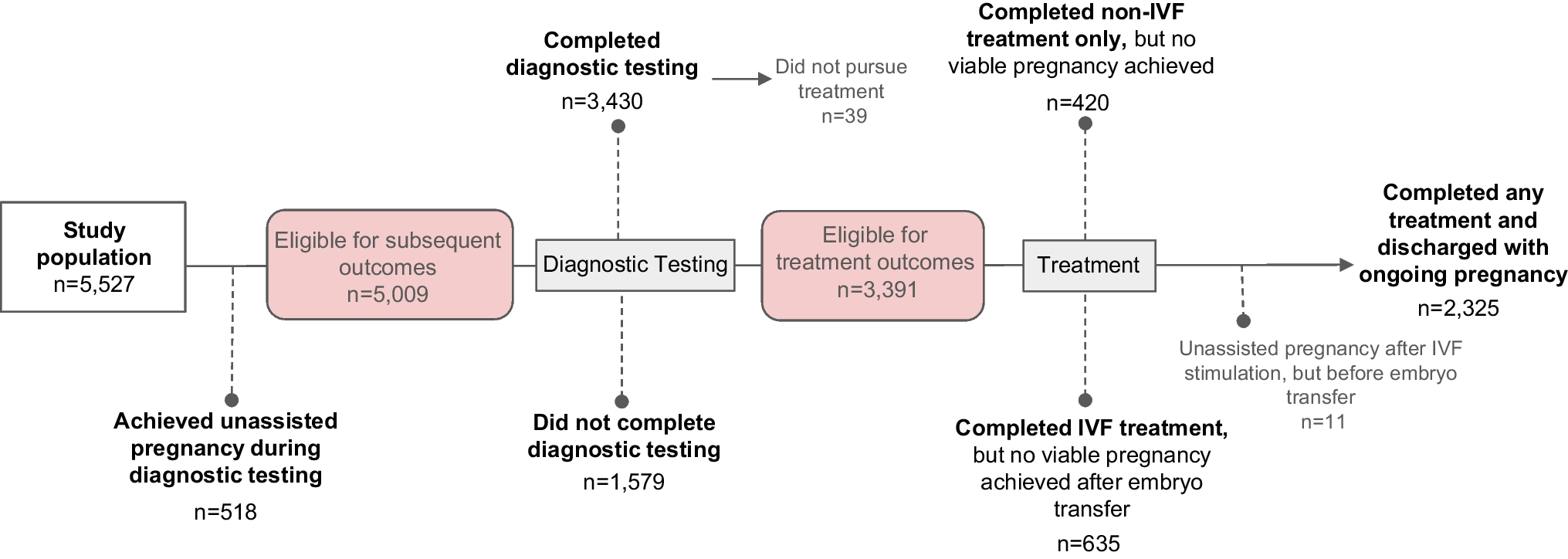
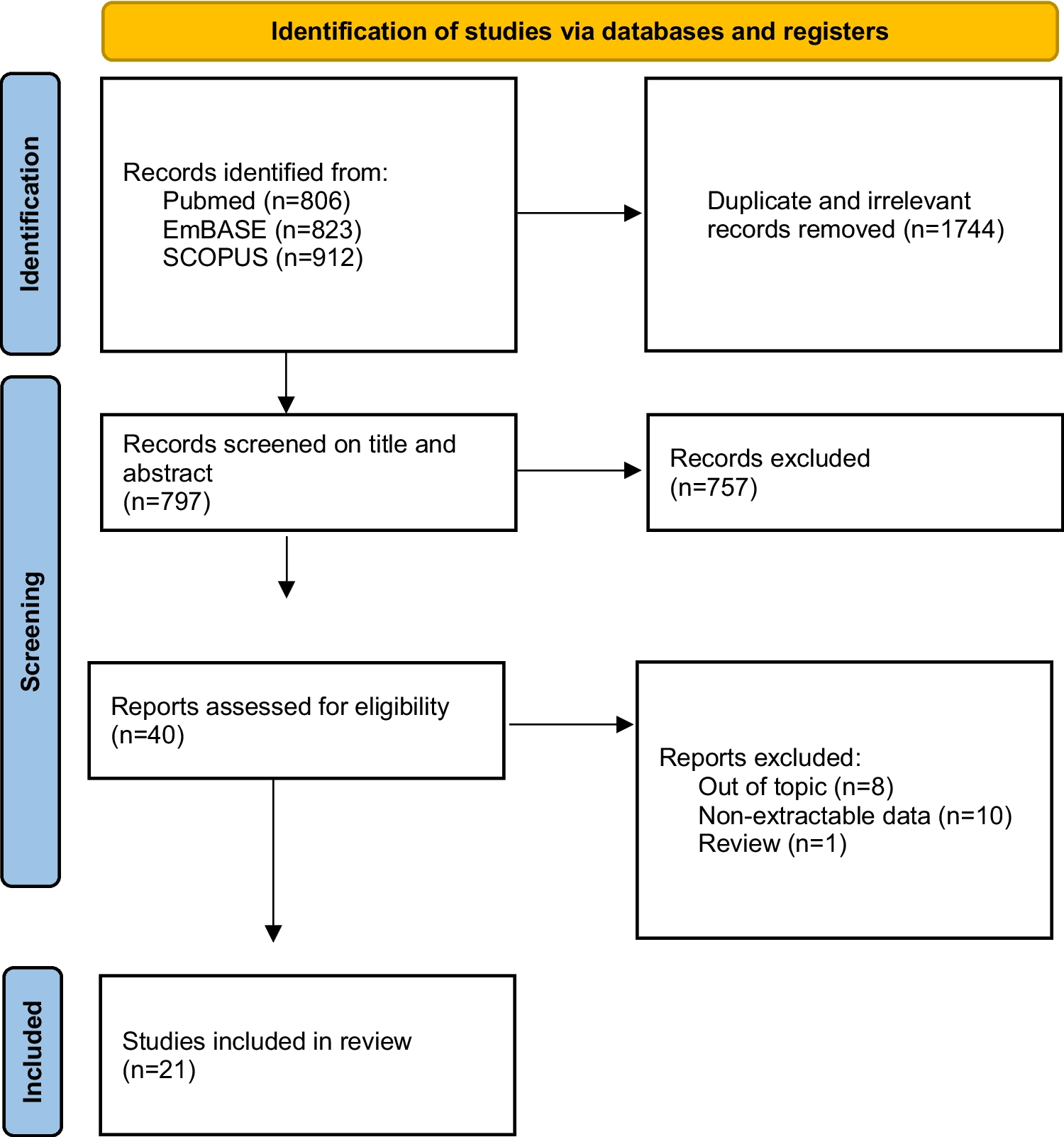
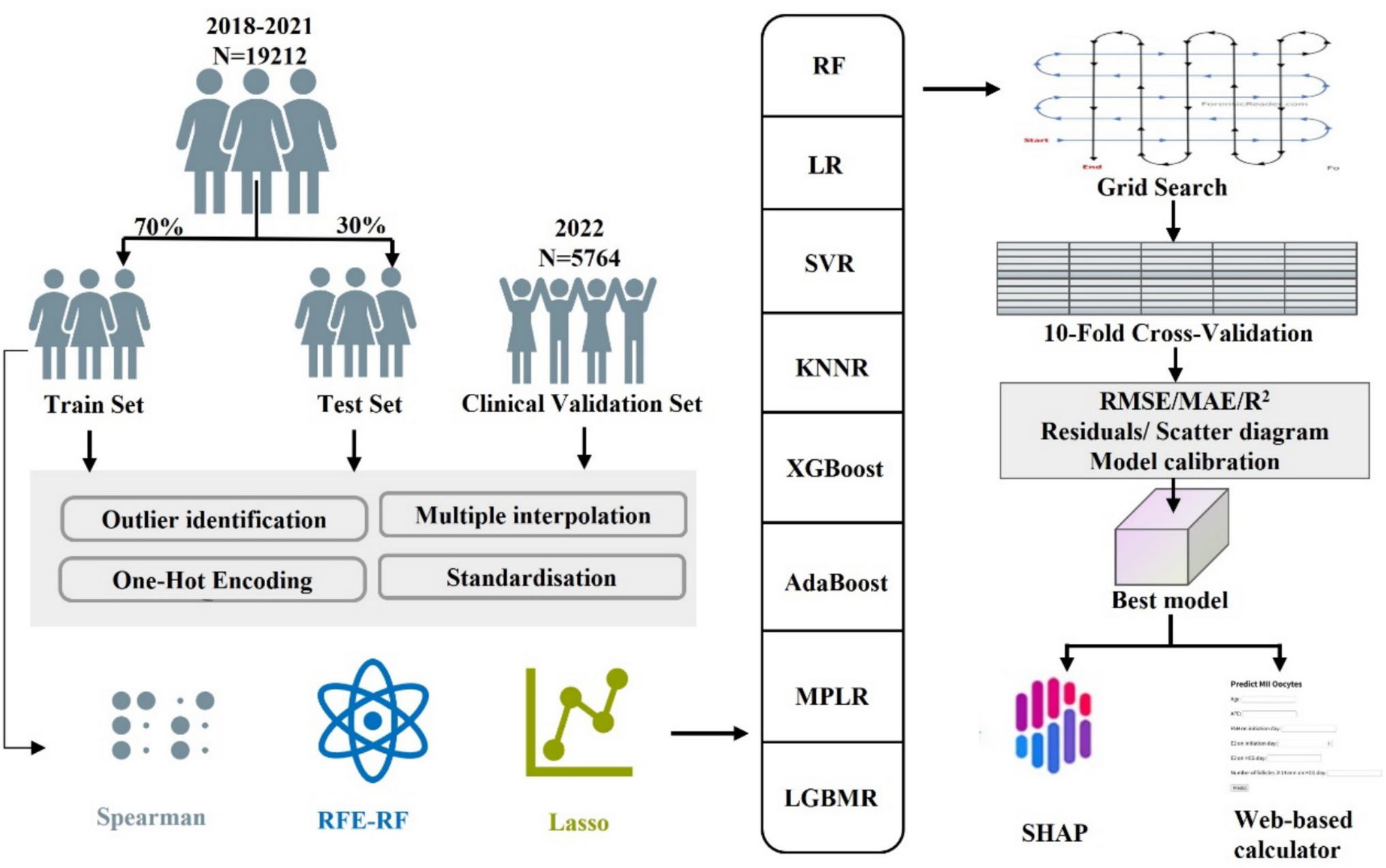
Remember me

Building on previous studies [11], our analysis using harmonic regression identified a robust 12-h (12-h) transcriptome across 46 human tissues, including the ovary, liver, adipose tissue, and muscle. We identified a total of 6912 genes exhibiting 24-h rhythmicity, 1855 genes with 12-h rhythmicity, and 974 genes displaying both 24-h and 12-h rhythms in at least one investigated tissue (Fig. 2A). Approximately 80% of these genes were protein-coding (Fig. 2B). Consistent with observations in murine and baboon tissues [3], approximately half of the 12-h REGs (52.51%) and a small proportion of the 24-h REGs (14.09%) exhibited oscillations with both 24-h and 12-h periods, depending on the tissue type (Fig. 2C).
Fig. 2
Identification of 12-h gene expression rhythms in human tissues. A, B Number of genes (A) and gene types in percent (B) for the total sets of 12-h REGs and 24-h REGs across 46 human tissues. Different colors indicate the gene types. C Number of 12-h REGs and 24-h REGs and their intersection across 46 human tissues (x-axis). The list of all 46 tissues studied and their corresponding short names can be found in Table S1. In panels A to C, all REGs in each tissue satisfy q(BH) < 0.2 and log2 peak-trough amplitude > 0.5. D Polar densities of DIPs (solid line) in males (dark blue) and females (light blue). E Number of 12-h REGs with an amplitude higher than a threshold (x-axis, log2) as a function of the threshold in all tissues combined for female (solid, light blue) and male (dashed, dark blue) donors. F Summary of the total number of 12-h REGs in each tissue (top), divided according to four statistical models for differential rhythmicity analysis (bottom): model 2 (blue), model 3 (cyan), model 4 (mustard), and model 5 (brick). See methods for details on the statistical models. M, male; F, female. In panels E and F, all REGs shown satisfy q(BH) < 0.2 and log2 peak-trough amplitude > 0.5 in one or both sexes (see methods)
Numerous studies have demonstrated sexually dimorphic patterns in 24-h gene expression rhythms, yet potential sex differences in 12-h transcriptomic rhythmicity remain largely unexplored. To investigate this, we analyzed transcriptome data from the GTEx project, stratified by sex. We applied harmonic regression with DIPs and a stringent inclusion criterion, considering only tissues with at least 24 samples in both sexes to minimize potential biases due to sample size variations. As a result, a total of 29 human tissues were included. Despite having comparable DIPs between males and females (Fig. 2D), females exhibited a higher number of 12-h REGs compared to males across all amplitude thresholds (Fig. 2E). Moreover, when considering peak-to-trough ratios exceeding 2, females exhibited approximately 100 more 12-h REGs than males (Fig. 2E). In the 29 common human tissues that both sexes have, although tissues such as stomach and transverse colon did not differ much, with most genes exhibiting statistically identical rhythms in the two groups (model 4 in Fig. 2F), the stratification by sex unveiled several highly dimorphic tissues (Fig. 2F). Notably, females had considerably more 12-h REGs in the liver, skin, and salivary gland (Fig. 2F). Taken together, our analysis demonstrated sex-dimorphic 12-h rhythmicity and an overall increased rhythmicity in females.
Midlife marks the onset of rhythmic gene expression reprogramming across human tissuesTo investigate how aging reprograms gene expression patterns, we conducted a comprehensive analysis across four age groups (20–39, 40–49, 50–59, and 60–69). To minimize potential biases arising from sample size variations in the harmonic regression analysis for rhythmic gene expression and the model selection for differential rhythmicity analysis, we applied a stringent inclusion criterion: only tissues with at least 24 samples in the age group with the smallest sample size were considered for further analysis. After this filtering, 30 human tissues were included in the study. To identify genes with age-related changes in rhythmicity, we defined differential rhythmicity genes as those classified under model 2 (rhythmic only in the younger group), model 3 (rhythmic only in the older group), or model 5 (rhythmic in both age groups but with divergent parameters). Our analysis identified differentially expressed genes (DEGs), differential 12-h REGs, and differential 24-h REGs by comparing the youngest group (20–39) to the others.
Although the DIP distributions were similar across all age groups (Fig. 3A), we observed that the reprogramming of 12-h and 24-h REGs began to manifest in samples from individuals aged 40–49 years. Most tissues showed a larger number of differential 24-h REGs than the number of DEGs and differential 12-h REGs (Fig. 3B to D). Notably, the ovary, prostate, visceral adipose tissue, sun-exposed skin, and colon sigmoid displayed distinct reprogramming of 12-h REGs in samples from individuals aged 40–49 years (Fig. 3B). In contrast, DEGs only started to emerge in samples from individuals aged 50–59 years (Fig. 3C).
Fig. 3
Rhythmic transcriptome analysis across 30 human tissues and four age groups. A Polar densities of DIPs in the 20–39 age group (sky blue), 40–49 age group (dark red), 50–59 age group (cobalt blue), and 60–69 age group (deep navy blue). B–D Number of genes for differential 12-h REGs, differential 24-h REGs, and DEGs between the 20 and 39 age group and the following age groups: 40–49 (B), 50–59 (C), and 60–69 (D). E, F Number of 24-h REGs (E) and 12-h REGs (F) with an amplitude higher than a threshold as a function of the threshold in all tissues combined for donors aged 20–39 (solid, light blue) and 40–49 (dashed, dark blue). G, H Summary of the total number of 24-h REGs (G) and 12-h REGs (H) in each tissue (top), divided according to four statistical models (bottom). Four statistical models for differential rhythmicity analysis (see methods) are represented: model 2 (blue), model 3 (cyan), model 4 (mustard), and model 5 (brick). Differential 12-h REGs are defined as 12-h REGs in models 2, 3, and 5. Differential 24-h REGs are defined as 24-h REGs in models 2, 3, and 5. Genes with |log2fold change (FC)|> 0.5 and adjusted p value < 0.05 are considered DEGs. Age groups: 20–39 and 40–49. All REGs shown satisfy q(BH) < 0.2 and log2 peak-trough amplitude > 0.5 in one or both age groups (see methods)
Therefore, we next focused on comparing individuals aged 40–49 with those aged 20–39. Individuals in the 40–49 age cohort showed modestly damped 24-h REGs (> 2 amplitude peak-to-trough ratio) (Fig. 3E). Most tissues, such as the breast, terminal ileum, and left ventricle of the heart, showed conserved 24-h rhythmicity across age (model 4 in Fig. 3G). However, certain tissues, including the transverse colon and coronary artery, demonstrated increased 24-h rhythmicity (model 3 in Fig. 3H). In contrast, 12-h REGs were amplified in the 40–49 age cohort (Fig. 3F), particularly in the ovary and sigmoid colon (model 2 in Fig. 3H).
In summary, our findings underscore the significance of midlife (40–49 years old) as a pivotal transition phase characterized by the onset of distinct rhythmic transcriptome reprogramming across human peripheral tissues.
Gene expression rhythms reprogramming in female reproductive systemOur analysis revealed that midlife, spanning ages 40 to 49, marks the onset of reprogramming in rhythmic gene expression across human tissues. This period is characterized by substantial hormonal fluctuations and physiological changes, particularly within the female reproductive system. Each component of the female reproductive system displayed distinct patterns of rhythmic gene reprogramming at various stages of aging: the ovary showed significant reprogramming of 12-h REGs during midlife (Fig. 3B and H), while the uterus experienced notable changes in 24-h REGs within the same age range (Fig. 3B and G). In contrast, the vagina exhibited marked reprogramming of 24-h REGs after age 50 (fig. S2, A and C).
Within the female reproductive system, the DIPs remained similar between the 20–29 and 40–49 age groups (Fig. 4A to C). In the vagina, the total number of 24-h REGs and their peak phases were similar between both age groups (Fig. 4A and D), with approximately 70% of genes displaying statistically identical 24-h rhythms (model 4 in Fig. 4G). However, when peak-to-trough ratios exceeded 1, the 24-h REGs were dampened in the older donors (Fig. 4D). In contrast, the ovary and uterus exhibited a higher number of total 24-h REGs in 40–49 age group (Fig. 4E and F). The uterus showed a significant enrichment of 24-h mRNA rhythms in individuals aged 40–49 at all amplitudes, characterized by an extensive evening wave (Fig. 4B and E), with about 65% of genes gaining 24-h rhythms (model 3 in Fig. 4H). The ovary exhibited a distinct pattern, with about 60% of genes showing statistically identical 24-h rhythms in both groups (model 4 in Fig. 3I). The remaining 24-h rhythms were either specific to one age group or displayed different rhythmic patterns between the two age groups (Fig. 4I).
Fig. 4
Characterization of age-associated alterations in 12-h and 24-h gene expression oscillations within the female reproductive system. A to C Polar densities of DIPs (solid line) in the vagina (A), uterus (B), and ovary (C) from donors aged 20–39 (light blue) and aged 40–49 (deep navy blue). D–F Number of 24-h REGs with an amplitude higher than a threshold as a function of the threshold in the vagina (D), uterus (E), and ovary (F) combined for donors aged 20–39 (solid, light blue) and 40–49 (dashed, dark blue). G to J Heatmaps of 24-h REGs and 12-h REGs in models 2 to 5 in each tissue: 24-h REGs in the vagina (G), uterus (H), and ovary (I); 12-h REGs in the ovary (J). Four statistical models for differential rhythmicity analysis (see methods) are represented: model 2 (blue), model 3 (cyan), model 4 (mustard), and model 5 (brick). Age groups: 20–39 and 40–49. Log2 (mean-centered) expression of all samples, from low (blue) to high (brown), represented by 1-h bins plotted with a 4-h moving average window. K, L Heatmaps of all genes in the vagina (K) and uterus (L), using the same color scale and representation as in G to J. All REGs shown satisfy q(BH) < 0.2 and log2 peak-trough amplitude > 0.5 in one or both age groups (see methods)
Remarkably, 12-h ultradian mRNA rhythms were notably detected in the ovary, with no statistically significant 12-h genes in the uterus or vagina (Fig. 4J to L). The ovary demonstrated a strong enrichment of 12-h mRNA rhythms in individuals aged 40–49 at all amplitudes (Fig. 4J and M). Taken together, these findings reveal tissue-specific changes in rhythmic gene expression within the female reproductive system during midlife, highlighting the ovary’s unique pattern of enriched 12-h rhythms, while the uterus and vagina exhibit distinct alterations in 24-h rhythms.
A comprehensive 12-h and 24-h transcriptome atlas of human ovary “middle-aging”We conducted a targeted analysis to investigate the distinctive reprogramming of ovarian gene expression rhythms in women aged 40–49. To ensure a precise and unbiased assessment of age-dependent changes in ovarian gene expression rhythms, stringent filtering criteria were applied to samples from both the 20–39 and 40–49 age groups. Samples showing histological evidence of premature ovarian aging (e.g., postmenopausal apoptosis in the 20–39 age group) or pathological changes (e.g., benign tumors in the 40–49 age group), as indicated by GTEx, were excluded from the analysis.
We observed a significant enrichment of 12-h mRNA rhythms across all amplitudes in individuals aged 40–49 (Fig. 5A). Additionally, there was a slight enrichment of 24-h mRNA rhythms across all amplitudes in this age group (Fig. 5B). Notably, the total number of differentially rhythmic genes (models 2, 3, and 5) exceeded the number of DEGs between age groups for both 12-h and 24-h rhythms (Fig. 5C). Intriguingly, downregulated genes in the 40–49 age group were enriched in rhythmic processes, and also in cell division, cell cycle regulation, and genomic stability, such as the G2/M checkpoint, mitotic cytoskeleton organization, DNA metabolic processes, and spindle localization (fig. S3A). The upregulated genes were primarily associated with myogenesis, the negative regulation of the intrinsic apoptotic signaling pathway in response to DNA damage, and epithelial-mesenchymal transition (fig. S3B).
Fig. 5
Molecular pathways linked to age-related changes in 12-h and 24-h transcriptome rhythms in the ovary. A, B Number of 12-h REGs (A) and 24-h REGs (B) with an amplitude higher than a threshold as a function of the threshold in the ovary combined for donors aged 20–39 (solid, light blue) and 40–49 (dashed, dark blue). C Number of DEGs, 24-h REGs, and 12-h REGs between the 20–39 and 40–49 age groups. The DEGs was divided into two subgroups: upregulated (up) and downregulated (down) genes. For differential rhythmicity analysis, 12-h REGs and 24-h REGs were divided into four categories based on statistical models: model 2 (blue), model 3 (cyan), model 4 (mustard), and model 5 (brick). See methods for details on the statistical models. Genes with |log2fold change (FC)|> 0.5 and adjusted p value < 0.05 were considered DEGs. D Top 20 clusters and their representative enriched terms (one per cluster) derived from Metascape-annotated pathways for genes exhibiting 12-h rhythmic expression exclusively in the 40–49 age group (model 3). E Thirteen clusters and their representative enriched terms (one per cluster) derived from Metascape-annotated pathways for genes exhibiting 12-h rhythmic expression with divergent parameters between the two age groups (model 5). F Histogram of peak phases for 12-h REGs from model 3 and model 5 in the 40–49 age group. G Heatmap of Metascape-annotated pathways for 12-h REGs from model 3 and model 5, grouped by peak phases in the 40–49 age group. In panels D to G, pathways enriched by over-representation analysis (ORA) (p < 0.05) are shown. Pathways not enriched are represented in grey. H, I PPI network (H) and MCODE components (I) identified using Metascape for 12-h REGs from model 3 and model 5, grouped by peak phases in the 40–49 age group. The resultant network contains the subset of proteins that form physical interactions with at least one other member in each group (H). Pathway and process enrichment analysis has been applied to each MCODE component independently, and the three best-scoring terms by p-value have been retained as the functional description of the corresponding components. Age groups: 20–39 and 40–49. All REGs shown satisfy q(BH) < 0.2 and log2 peak-trough amplitude > 0.5 in one or both age groups (see methods)
Moreover, the 12-h rhythms exhibited a greater number of differentially expressed genes—817 in model 3, 67 in model 5, and 3 in model 2—compared to the 24-h rhythms (Fig. 5C). The gained 12-h genes in individuals aged 40–49 (model 3) were predominantly enriched in angiogenesis-related processes, including vasculature development, actin cytoskeleton organization, VEGFA-VEGFR2 signaling, cytoskeleton in muscle cells, regulation of cell-substrate adhesion, positive regulation of cell migration, regulation of actin filament-based processes, response to growth factors, and negative regulation of cell adhesion (Fig. 5D). Additionally, the analysis revealed 67 genes exhibiting altered oscillation amplitudes or phase shifts between the two age groups (model 5 in Fig. 5C), which involved in cellular metabolism and homeostasis, including ribonucleoside catabolism, response to nutrient levels, protein processing in the endoplasmic reticulum, and lysosomal organization (Fig. 5E).
We further analyzed the differential 12-h REGs identified in models 3 and 5, comparing the 40–49 age group to the young group. These genes exhibited two distinct peak expression patterns: morning/evening and afternoon/night (Fig. 5F). To elucidate the biological processes associated with these differentially 12-h REGs, we categorized them based on their peak expression times in the 40–49 group and performed pathway enrichment analysis on each subset. Intriguingly, angiogenesis processes were predominantly enriched in genes peaking from 12 to 2 AM, cilium development pathways were enriched in genes peaking from 6 to 7 AM, and protein synthesis pathways were enriched in genes peaking from 11 AM to 12 PM (Fig. 5G).
To investigate the direct interactions between proteins encoded by differential 12-h REGs, we performed PPI enrichment analysis on each subsets. The best-scoring genes identified were involved in vasculature development, blood vessel development, and tube morphogenesis, emphasizing the involvement of angiogenesis in differential 12-h REGs peaking from 0 to 2 AM (Fig. 5H). MCODE method was subsequently applied to identify closely related proteins from the PPI network, resulting in the identification of nine subclusters. Remarkably, the functional annotations enriched in the three largest clusters were all related to angiogenesis-related processes, including positive regulation of chemotaxis, focal adhesion, and actin filament-based processes. The proteins in these three largest clusters belonged to the set of differential 12-h REGs peaking from 12 to 2 AM. In contrast, the smaller cluster belonged to the set of differential 12-h REGs peaking from 6 to 7 AM was composed of microtubule-associated proteins such as CEPB43, CPEB290, OFD1, HAUS5, and TEME216. The clusters belonged to the set of differential 12-h REGs peaking from 11 AM to 12 PM were composed of proteins associated with protein processing in the endoplasmic reticulum, SRP-dependent cotranslational protein targeting to the membrane, and RHOB and RHOC GTPase cycles.
Only three genes (FCGBP, MS4A4A, and PDK4) showed a loss of 12-h rhythmicity in the same age group (model 2 in Fig. 5C). Notably, these three genes showed no significant differential expression between the two age groups (fig. S3C). Furthermore, our analysis identified 77 genes exhibiting shared 12-h rhythms between the two age groups (model 4 in Fig. 5C), which were enriched in cell-substrate adhesion, enzyme-linked receptor protein signaling pathway, L1CAM interactions, cell migration involved in sprouting angiogenesis, RAF-independent MAPK1/3 activation, response to extracellular stimulus, the regulation of transforming growth factor beta (TGF-β) production, extracellular matrix disassembly, and TGF-β receptor signaling that activates SMADs (fig. S3D).
Although the 24-h rhythmic changes in the ovaries of middle-aged women are not as pronounced as the 12-h changes, we still carried out an analysis of differentially 24-h REGs, with the results shown in the supplementary figures. In brief, genes adopted de novo 24-h rhythmicity (model 3) in the 40–49 age group were enriched in both adaptive immune responses and angiogenesis-related processes (fig. S3E). Moreover, 89 genes with altered oscillation amplitudes or phase shifts (model 5) between the two groups were involved in the aromatic compound catabolic process, the Fas ligand pathway, and the stress induction of heat shock and cellular detoxification (fig. S3F). Additionally, 29 genes that lost 24-h rhythmicity (model 2) in individuals aged 40–49 were primarily associated with immune cell signaling and activation (Fig. S3G), particularly in macrophages, including NKRF, VSIG4, TYROBP, MPEG1, HSPE1, MRO, and NOTCH2. Furthermore, 880 genes exhibiting shared 24-h rhythms (model 4) between the two groups (Fig. 5C) were enriched in mTORC1 signaling, lipid biosynthetic processes, TNFA signaling via NFKB, and ferroptosis (fig. S3H).
Aging amplified the 12-h oscillatory expression patterns of angiogensis in the ovaryOur analysis revealed that, regardless of the rhythm analysis period (12 or 24 h), when contrasted with those in the 20–39 age group, individuals aged 40–49 exhibited a greater gain in rhythmicity (model 3) compared to both the loss of rhythmicity (model 2) and the changes in oscillation amplitudes or phase shifts (Fig. 5C). Importantly, gene ontology analysis revealed that both subsets of genes that gained 24-h and 12-h rhythmicity in the 40–49 age group were significantly enriched in angiogenesis-related processes (Fig. 5D and fig. S3D). Therefore, we expanded on results to determine the significant biological processes implicated in the 12-h and 24-h REGs in the 40–49 and 20–39 age group. In the 20–39 age group, only 24-h REGs were associated with the enrichment of these processes, whereas in the 40–49 age group, angiogenesis-related processes were enriched by both 12-h and 24-h REGs (Fig. 6A).
Fig. 6
Molecular pathways linked to age-induced 12-h and dual rhythmic expression of novel genes in the ovary. A Heatmap of Metascape-annotated pathways for 12-h REGs and 24-h REGs in the 20–29 and 40–49 age groups. Pathways enriched by over-representation analysis (ORA) (p < 0.05) are shown. Pathways not enriched are represented in grey. B Venn diagrams displayed the overlap between the 12-REGs and 24-REGs in the 20–29 (lower panel) and 40–49 (upper panel) age groups. The numbers indicate the number of genes in each section of the diagram. C Top 20 clusters and their representative enriched terms (one per cluster) derived from Metascape-annotated pathways for 12-h REGs, 24-h REGs, and their intersection in the 40–49 age group. D Venn diagrams displayed the overlap among the intersection of 12-h REGs and 24-h REGs in the 40–49 age group, 24-h REGs and 12-h REGs in the 20–29 age group. E Fourteen clusters and their representative enriched terms (one per cluster) were derived from 109 genes in D that exhibited dual rhythmicity (i.e., 12 h and 24 h) in the 40–49 age group and had previously been classified as 24-h REGs in the 20–29 age group. F Metascape visualization of the enrichment network showing the intracluster and intercluster similarities of enriched terms in (E). Each term is represented by a circular node, the color of which represents the cluster identity (i.e., nodes of the same color belong to the same cluster). Pathways enriched by over-representation analysis (ORA) (p < 0.05) are shown. All REGs shown satisfy q(BH) < 0.2 and log2 peak-trough amplitude > 0.5 in one or both age groups (see methods)
Interestingly, the 40–49 age group exhibited not only an increased number of 12-h REGs but also a significant rise in the ratio of dual rhythmicity genes relative to both the 12-h REGs (24.32%, 188/773) and 24-h REGs (16.27%, 188/1155). Further analysis revealed that all three gene sets—12-h REGs, 24-h REGs, and dual rhythmicity genes—in the 40–49 age cohort were linked to angiogenesis processes (Fig. 6C). Notably, pathways specifically involved in endothelial cell migration during angiogenesis, such as cell-substrate adhesion, regulation of cell-substrate adhesion, and cell–cell adhesion, were enriched exclusively in the 12-h REGs (Fig. 6C).
To investigate the origin of the increased number of dual rhythmicity genes in the 40–49 age group, we compared the genes exhibiting both 12-h and 24-h rhythms in this group to the corresponding 12-h and 24-h REGs in the younger cohort (20–39 years). Notably, about 58% (109 out of 188) of the dual rhythmicity genes identified in the 40–49 age group had previously been classified as 24-h REGs in the younger cohort (Fig. 6D). Pathway analysis of these 109 dual rhythmicity genes revealed significant enrichment in pathways related to angiogenesis, actin cytoskeleton regulation, positive regulation of apoptosis, mitochondrial organization, and PI5P, PP2A, and IER3 regulation of the PI3K/AKT signaling pathway (Fig. 6E). Network analysis further demonstrated that the angiogenesis pathway was closely connected to the regulation of peptidyl-tyrosine phosphorylation, positive regulation of protein modification processes, PI5P, PP2A, and IER3 regulation of PI3K/AKT signaling, actin cytoskeleton regulation, inflammatory responses, and focal adhesion (Fig. 6F).
Comments (0)