
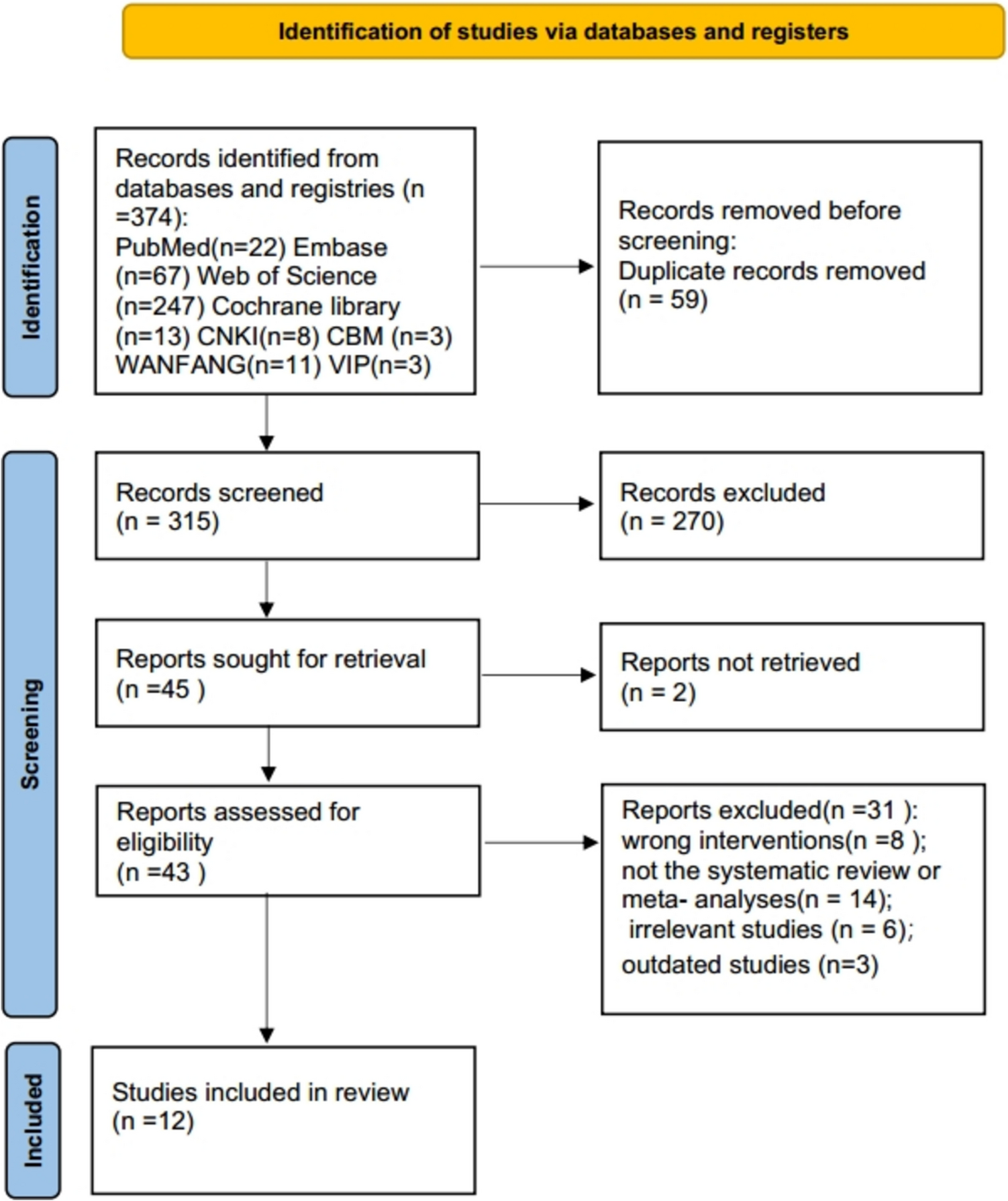
Remember me





To construct a panoramic map of necroptosis-related genes in all samples, the expression profiles from the GSE34526, GSE8157, and GSE5090 datasets were integrated. Since the data sets from diverse resources usually had serious batch effects, the raw data were corrected for batch effects and log normalized. The results showed (Fig. 1A, B) that the expression distributions of all samples were consistent after batch correction and log normalization; thus, this process improved the accuracy and robustness of the downstream analysis. Then, the PCA diagram (Fig. 1C, D) and UMAP diagram (Fig. 1E, F) were drawn to show the sample distribution of the original data before as well as after the correction of the batch effect.
Fig. 1
Panorama of necroptosis genes in PCOS. A Expression distribution chart before batch effect correction. B Expression distribution chart after batch effect correction; samples are plotted on the x-axis, while expressions are on the y-axis, and a box line in the figure shows the distribution of the expression in the sample. C PCA sample distribution chart before batch effect correction. D PCA sample distribution chart after batch effect correction. E UMAP sample distribution chart before batch effect correction. F UMAP sample distribution chart after batch effect correction. G Circular map of chromosome localization. The outermost circle is the chromosome and the position of the gene on the chromosome, and the second circle is the positioning of the chromosome corresponding to the gene. H Heatmap. The row is the necroptosis gene, and the column is the sample. The samples in different data sets or groups are marked with different color blocks. In the figure, red and blue represent high- and low-expression values, respectively
Next, a panoramic view of gene positioning on chromosomes was drawn based on the positioning of these genes in chromosomes and combined with various circular display modes (Fig. 1G). The results showed that the chromosomal locations of some genes were very close, indicating that those genes are closely related at the genomic level and might have similar expression characteristics at the transcriptome level. In addition, heatmaps were used to visualize the similarities and differences in necroptosis-related gene expression levels between the normal and disease groups in the three data sets (Fig. 1H). The results showed that in the three data sets, some genes related to necroptosis had different expression trends.
Expression and correlation analysis of necroptosis-related genes in polycystic ovary syndromeThe expression levels of necroptosis-related genes in the three data sets and the merged datasets were extracted for PCA. The results showed that the expression of necroptosis-related genes in the GSE34526 data set was better at distinguishing normal and PCOS (Fig. 2A), but the expression of necroptosis-related genes in GSE8157 (Fig. 2B), GSE5090 (Fig. 2C), and merged data set (Fig. 2D) could not clearly distinguish normal and PCOS samples. The source of the GSE34526 data set was ovarian granulosa cells, which indicates that the expression of PCOS is greatly affected by sampling. Finally, Pearson’s correlation analysis was performed using the merged data set, and correlation analysis results were illustrated by constructing a correlation heat map (Fig. 2E). The results showed that there was a strong correlation between necroptosis-related genes.
Fig. 2
Analysis of expression and correlation of necroptosis-related genes in PCOS. A PCA of necroptosis gene expression in GSE34526 data set. B PCA of necroptosis gene expression in GSE38157 data set. C PCA of necroptosis gene expression in GSE5090 data set. D PCA of necroptosis gene expression in merged data sets. E Correlation heatmap. Blue indicates a positive correlation, and red indicates a negative correlation. The larger the proportion of the pie chart in the square of the figure, the larger the absolute value of the intergenic correlation corresponding to the x-axis and y-axis
The necroptosis-related gene-based diagnosis modelBecause the expression of necroptosis genes has important biological significance, a diagnostic model of PCOS was constructed based on all necroptosis genes and genes with significant differences with an absolute value of more than 0.7 (p < 0.05). First, genes were selected by univariate logistic regression (Fig. 3A) and further identified by a tenfold cross-test (Fig. 3B). Finally, 25 statistically significant (p < 0.05) genes were selected. Subsequently, a forest map was used to visualize the diagnostic model (Fig. 3C). The results showed that the base with the largest absolute value of the influence coefficient in this model was atg9a, which is a necroptosis gene, and the influence coefficient was 2.97.
Fig. 3
Constructing a diagnostic model of necroptosis-related genes. A This figure shows the convergence-identifying process of logistic regression for 25 gene features. The L1 Norm value is plotted on the x-axis, the regression coefficient on the y-axis, and the lines of different colors represent different features. B tenfold cross-inspection chart and Lambda value selection curve. This figure is used to select the best lambda value of the logistic regression model. Generally, the lowest point is selected; that is, the dotted line on the left side of the figure is the best lambda value. C Forest diagram of the diagnostic model. In the first column are the 25 genes used in model making, while the second, as well as the corresponding graph, represents influence coefficients associated with each of these genes in the model, and the third column is the significance index p-value of these genes
Subsequently, for verifying the model’s accuracy, a logistic multivariate model including the above 25 genes (CD9, amd1, psmd3, CTNNAL1, atg9a, SOCS2, nfkbie, LMO2, gstm2, ncf4, Cbln1, ptpn6, Blnk, linc00597, LGALS2, man2b1, stxbp2, Ncf1, dsg2, Tacc3, map1s, atp13a2, HAMP, sync, pycard) was constructed, and it was visualized by nomogram (Fig. 4A). After identifying by multivariate logistic regression, the results showed that 10 genes (amd1, CTNNAL1, atg9a, nfkbie, gstm2, ptpn6, Blnk, linc00597, HAMP, pycard) had a greater impact on the prediction model. After that, the model’s prediction efficiency has been further tested utilizing the recall curve as well as the DAC curve (Fig. 4B, C). The results show that the model has superior prediction efficiency and robustness in the three verification methods. Subsequent analysis was mainly based on these 10 genes.
Fig. 4
Validation of diagnostic models. A Nomogram, the left side is the predictor and the right is the ruler. B DCA curve, the x-axis represents the risk threshold, and the y-axis represents the net benefit rate, the Treat None (orange) represents the 0 net benefit rate, the Treat All (red) represents that all samples have received the intervention, and the other color lines represent the curves corresponding to the genes in the model. C Calibration curve. By plotting the fitting of the actual probability and the predicted probability of the model under different conditions in the figure, the curve can be well fitted to the diagonal, and the model has a good prediction effect on the actual results
Network analysis of hub genesSince there are universal relationships among genes and genes regulating the same biological process will have closer relationships, hence for further analyzing the interactions among hub genes, the PPI networks were constructed using the GeneMANIA plug-in of Cytoscape software for the following 10 hub genes (AMD1, CTNNAL1, ATG9A, NFKBIE, GSTM2, PTPN6, BLNK, LINC00597, HAMP, PYCARD). We have subsequently visualized them using Cytoscape software (Fig. 5A). Cytohubba was used to differentially analyze the PPI network to obtain the core network of top10 MCC scores, in which GSTM2, PTPN6, NFKBIE, and BLNK were hub genes selected by previous diagnostic models(Fig. 5B).
Fig. 5
Network analysis of hub genes. A PPI network. The black shaded dots are hub genes, and the others are genes highly related to hub genes. B The core network of top10 genes scored by MCC. C miRNA gene regulatory network, yellow are miRNAs, and the rest are hub genes. D Transcription factors—gene regulatory network. Blue represents transcription factors, and the rest are hub genes. E Drugs—gene regulatory network. Purple represents the drug names, and the rest are hub genes. F RBP—gene regulatory network. Yellow represents RBP, and the rest are hub genes
Using the CyTargetLinker plug-in, microRNA-related link sets were selected for miRNA prediction, and the miRNA gene regulatory network was obtained (Fig. 5C). TF-related link sets were selected for transcription factor prediction to obtain the transcription factor gene regulatory network (Fig. 5D). Drug-related link sets were selected for transcription factor prediction to obtain the drug-gene regulatory network (Fig. 5E). RBP prediction was performed using the starBase database, and the RBP gene regulatory network was obtained (Fig. 5F). The results show that these genes not only have exclusive miRNAs, transcription factors, RBPs, or drugs but also share the same phenomenon indicating that they may be subjected to the same regulatory process to reflect similar biological functions.
Unsupervised clustering of samplesThe previously selected 10 genes can be used to distinguish samples with different disease states, so the expression profiles of the 10 genes were extracted to perform unsupervised clustering on all samples using the PAM method in consistent clustering. First, the optimal number of clusters was determined by the elbow graph and consistency matrix (Fig. 6A–J). According to the results, when the clusters were 3 in number, the heatmap of the consistency matrix showed a clear sample distribution, while the scatter points on corresponding positions of the elbow map were also at the peak position. Therefore, k = 3 was chosen as the optimum cluster number for unsupervised clustering where all samples were clustered into three categories.
Fig. 6
Molecular typing of unsupervised samples. A The consistent cluster cumulative distribution function (CDF) when k = 2–9. B Elbow diagram. Relative change of area under CDF curve when k = 2–9. C–J Heatmap of consistency matrix when k = 2–9. K Legends of C–J when k = 2–9
By clustering the samples in the diseased state using the expression profiles of all the significantly different genes, it was also found that the best number of clusters was 3 (Fig. 7A–C). Comparing the two clustering results, it was found that most of the samples included in category 1 were the same, except that the samples in categories 2 and 3 were quite different, which indicates that the selection of 10 genes using clinical models was very representative. Finally, the clustering results in Fig. 6D were used as the final molecular typing results of the PCOS samples.
Fig. 7
Unsupervised sample molecular typing. A Consistency clustering CDF when k = 2–9. B Elbow diagram. Relative change of area under CDF curve when k = 2–9. C Heatmap of consistency matrix when k = 3
GO/KEGG enrichment analysisTo analyze the biological processes or functions affected by DEGs between normal samples and disease samples, GO and KEGG enrichment analyses were performed on all DEGs (Fig. 8A–D). GO enrichment analysis enriched 419 GO terms in total, including 360 BP terms which were mainly related to myeloid leukocyte activation, maintenance of location, and regulation of cation transmembrane transport; 39 MF classes being representative of GO terms included SH2 domain binding and glutathione binding and 20 CC classes. The representative GO terms included alpha–beta T-cell receptor complex and apical cortex. A total of 9 pathways were enriched by KEGG enrichment analysis where four pathways with the highest number of enriched genes were the B-cell receptor signaling pathway, PD-L1 expression, and PD-1 checkpoint pathway in cancer, T-cell receptor signaling pathway, and pathogenic Escherichia coli infection.
Fig. 8
GO/KEGG enrichment analysis. A Bar graph of BP classification enrichment results of GO. The x-axis is the number of genes enriched on the GO term, the color corresponds to the − log10 value (p-value), and the y-axis is the enriched GO term. B Network diagram of CC classification and enrichment results of GO. The number of genes enriched to the GO term is represented by the size of the dot, and a connecting line indicates that the gene is enriched to the corresponding GO term. C Bubble diagram of MF classification and enrichment results of GO. The x-axis is the percentage of genes enriched to go terms, the color corresponds to the − log10 value (p-value), and the y-axis is the enriched go terms. D KEGG enrichment result network diagram. The number of genes enriched into the KEGG pathway is represented by the size of the dot, while color corresponds to the − log10 value (p-value)
GSEA/GSVA enrichment analysis of high as well as low necroptosis groupsFor evaluating the correlation between BPs or functions of necroptosis genes in PCOS diseases, the expression profiles of all disease samples were extracted, and then, disease samples were grouped according to the median expression of the ATG9A gene. Limma package was used for differential analysis, and the results of the differential analysis were used for GSEA enrichment analysis and GSVA analysis. A total of 960 biological pathways were enriched by GSEA enrichment analysis, and 11 significantly different pathways were obtained by GSVA analysis. The ridge map was used to show the enrichment scores and significance p-values of these 11 pathways (Fig. 9A). The volcano map (Fig. 9B) was used to show the results of pathway difference analysis, and the heatmap (Fig. 9C) shows the scores of pathways with significant difference analysis in different samples.
Fig. 9
GSEA and GSVA enrichment analysis. A The mountain map of GSEA analysis results. The x-axis is the enrichment score of the enriched term, and the color corresponds to the corrected p-value. B The volcano diagram where the x-axis is the difference multiple after taking log2, the y-axis is − log10 (p-value), and the blue and red are significantly down- and up-regulated pathways, respectively. C The hot map of pathway enrichment scoring. The upper color bar represents the data set of the sample source; each color represents a data set: blue represents a low score, and red represents a high score
Immune infiltration analysisThe enrichment analysis results showed that there were significant differences in the immune process between the two groups of samples. Therefore, to further analyze the immune infiltration level between the two groups of samples, the scores of 28 immune cells for all samples were calculated using the ssGSEA method. The results showed that all 10 hub genes were significantly correlated with Th2 cells (Fig. 10A). A correlation scatter plot was used for visualization of the correlation significance test (Fig. 10B–H). The results showed that many of the 28 types of immune cells, including cytotoxic cells, macrophages, NK CD56bright cells, and Th17 cells, showed significant differences between the two groups of samples. In addition, the correlation between hub genes and immune cell infiltration levels shows that the BLNK gene has a strong positive correlation with macrophages, indicating that this gene has important research value.
Fig. 10
Correlation analysis between hub genes expression and immune characteristics of ssGSEA. A Lollipop chart of the correlation between Th2 cell scores and 10 hub genes. B Correlation scatter plot among cytotoxic cells and PYCARD gene. C Correlation scatter plot among macrophage cells and BLNK gene. D Correlation scatter plot among NK CD56bright cells and NFKBIE gene. E Correlation scatter plot among NK CD56bright cells and PTPN6 gene. F Correlation scatter plot among NK CD56bright cells and HAMP gene. G Correlation scatter plot among Th17 cells and CTNNAL1 gene. H Correlation scatter plot among Th17 cells and ATG9A gene
We used cybersport as another method for calculating the immune infiltration level of 22 immune cells in the samples (Fig. 11A–E). Similarly, the correlation between 10 hub genes and immune cell infiltration level has been evaluated, and thereby, significantly related gene immune cell pairs were selected. The results showed a multiple positive correlation like Tregs with HAMP, monocyte cells with NFKBIE, macrophage M2 cells with BLNK, macrophage M2 cells with PYCARD, and mast-activated cells with NFKBIE.
Fig. 11
Correlation and the difference between the expression of hub genes and CIBERSORT immune characteristics. A Correlation scatter plot among regulatory T (Treg) cells and HAMP gene. B Correlation scatter plot among monocyte cells and NFKBIE gene. C Correlation scatter plot among macrophage M2 cell enrichment score and BLNK gene. D Correlation scatter plot among macrophage M2 cell enrichment score and PYCARD gene. E Correlation scatter plot among mast-activated cells and NFKBIE gene (each point in the A–E diagram represents a sample, a straight line is the correlation fitting curve, and the shaded part is the confidence). F Grouping comparison diagram of regulatory T (Treg) cell enrichment scores among high as well as low groups of HAMP expression. G Grouping comparison chart of monocyte enrichment scores among high as well as low groups of NFKBIE expression. H Group comparison diagram of macrophage M2 cell enrichment scores among high as well as low groups of BLNK expression. I Group comparison diagram of macrophage M2 cell enrichment scores among high as well as low groups of PYCARD expression. Student’s t-test was used for the difference between groups. The difference with statistical significance was indicated by “*,” where “*” indicates p < 0.05, “* *” indicates p < 0.01, “* * *” indicates p < 0.001, “* * * *” indicates p < 0.0001, and “ns” indicates insignificant
Finally, boxplots were used for visualization and statistical significance testing (Fig. 11F–I). The results showed that the enrichment scores of regulatory T cells were considerably different among high- and low-expression groups of HAMP (Fig. 11F), the enrichment scores of monocytes were considerably different among the high- and low-expression groups of NFKBIE (Fig. 11G), and the enrichment scores of macrophage M2 cells were considerably different among the high- and low-expression groups of BLNK and PYCARD (Fig. 11H, I).
Correlation and difference between hub genes and molecular subtypesAccording to the results of molecular typing of diseased samples using necroptosis gene expression before, Wilcox’s test has been utilized for comparing the differences in the hub gene expression among three molecular typing samples. There were significant differences in nine genes (CTNNAL1, ATG9A, NFKBIE, GSTM2, PTPN6, BLNK, LINC00597, HAMP, PYCARD). Box plots were used for visualization and statistical significance tests (Fig. 12A–I) and for visualizing and testing the significance of correlations using correlation scatter plots (Fig. 12J–L). The results showed that there were three genes significantly related to the molecular typing grouping: BLNK, LINC00597, and PYCARD.
Fig. 12
Correlation and difference between the expression of molecular subtypes and hub genes in samples. Box plot of CTNNAL1 (A), ATG9A (B), NFKBIE (C), GSTM2 (D), PTPN6 (E), BLNK (F), LINC00597 (G), HAMP (H), and PYCARD (I) in molecular typing grouping. Scatter plot of correlation between BLNK (J), LINC00597 (K), PYCARD (L), and molecular typing. Wilcox’s test has been utilized to analyze the differences between groups. The difference with statistical significance was indicated by “*” where “*” indicates p < 0.05, “* *” indicates p < 0.01, “* * *” indicates p < 0.001, “* * * *” indicates p < 0.0001, “ns” indicates not significant, and “ns.” means p = 1
A correlation scatter plot has been utilized to show the correlation between molecular typing and immune cells. The results showed that cytotoxic cells (R = 0.67, p = 0.00018), iDC (R = 0.46, p = 0.017), macrophages (R = 0.62, p = 0.00072), NK CD56dim cells (R = 0.58, p = 0.0019), NK cells (R = − 0.52, p = 0.0065), T cells (R = 0.65, p = 0.00036), helper T cells (R = 0.53, p = 0.0056), Th2 cells (R = − 0.47, p = 0.015), T-cell CD4 memory resting (R = 0.63, p = 0.00061), regulatory T cells (Tregs) (R = − 0.44, p = 0.025), macrophage M2 (R = 0.55, p = 0.0034), and mast resting cells (R = 0.48, p = 0.013) were significantly correlated with molecular typing (Fig. 13A–L).
Fig. 13
Correlation between the expression of molecular subtypes in samples and immune infiltration. Scatter plot of correlation between cytotoxic cells (A), iDC (B), macrophages (C), NK CD56dim cells (D), NK cells (E), T cells (F), helper T cells (G), Th2 cells (H), CD4 memory resting T cells (I), regulatory T cells (Tregs) (J), macrophage M2 (K), mast resting cells (L), and molecular subtype grouping of samples. Wilcox’s test has been utilized for analyzing the differences between groups. The difference with statistical significance was indicated by “*,” where “*” represents p < 0.05, “* *” for p < 0.01, “* * *” for p < 0.001, “* * * *” for p < 0.0001, and “ns” means not significant
Comments (0)