Cell lines
Human Embryonic Kidney cells (HEK293T) (ATCC® CRL-3216™) cells and African green monkey kidney (Vero) (ATCC® CCL-81 ™) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Fisher Scientific, Gibco™) with 10% FBS (Thermo Fisher Scientific, Gibco™) and 2% Penicillin/Streptomycin (Invitrogen, Inc., Carlsbad, CA).
Reagents and antibodies
Curcumin (Cur), Gallic acid (GA), Quercetin (Q), Silymarin (Sil), (3-aminopropyl) triethoxysilane (APTES, ≥ 98%), EDC N-(3-dimethylaminopropyl)-N0-ethylcarbodiimide hydrochloride, and N-hydroxy succinimide (NHS) were all purchased from Sigma-Aldrich, USA. MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) dye and dimethyl sulfoxide (DMSO) were purchased from Serva Electrophoresis GmbH, Germany. Lipofectamine 3000 reagent (Thermo Fisher Scientific, Invitrogen™) was used for transfection. Spike protein ectodomain encoding plasmid, a generous gift from Dr. Jason S. McLellan (Wrapp et al. 2019), was purified with PureLink™ HiPure Plasmid Maxiprep Kit (K210006, Thermo Fisher Scientific, Invitrogen™). The antibodies used were as follows: Rabbit anti-spike antibody (40,591-T62, Sino biological), spike monoclonal antibody (E-AB-V1008, Elabscience), Goat anti-rabbit antibody (ab6721, Abcam), and Goat anti-human IgG FITC (E-AB-1019, Elabscience). Protein production and quantification were performed by Strep-Tactin®XT Spin Column (2-4151-000, iba-lifesciences) and BCA assay kit (23,225, Pierce). Recombinant human ACE2 (PKSH032068, Elabscience) and Recombinant 2019-nCoV spike (PKSR030477, Elabscience) were used in drug screening.
Plasmid preparation and transfection
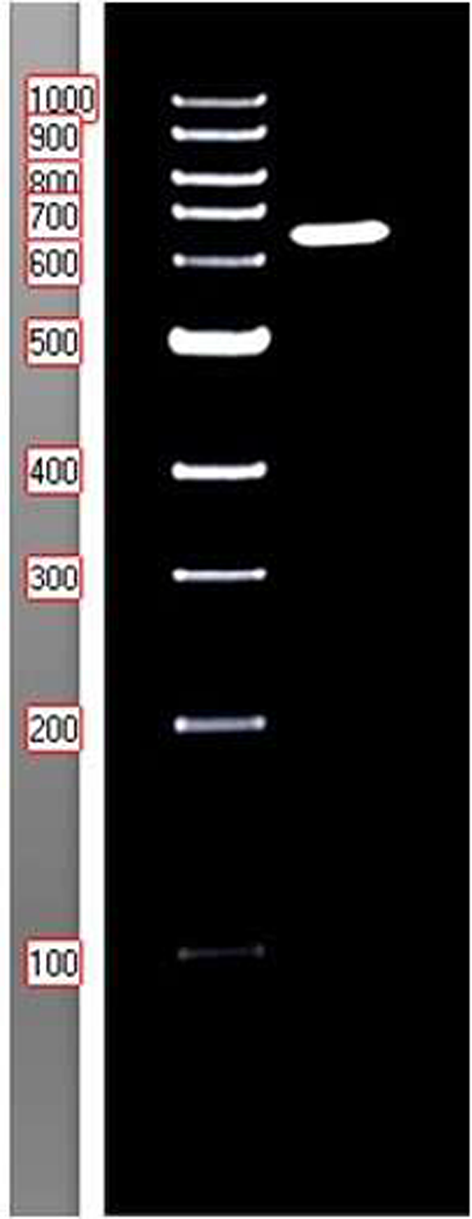
The plasmid expressing C-terminal Twin strip tagged spike protein was amplified and purified with a maxiprep kit following the manufacturer’s instructions. HEK293T cells were seeded in a 6-well plate, 6 × 105 cells per well, in DMEM complete media. The following day, the cells were transfected with the plasmid using Lipofectamine 3000 reagent, following the manufacturer protocol. The cells were harvested 72 h post-transfection and prepared for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis.
Western blot
For the preparation of cell lysates, cells were washed twice with ice-cold PBS, harvested, re-suspended in Laemmli’s buffer, and boiled for 5 min. Samples were separated on 9% SDS‐PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes (Amersham). The membrane was then blocked with 5% non-fat dry milk in TBS and incubated with primary antibody (1:1000) overnight at 4 °C. Following incubation with HRP‐labelled secondary antibody (1:1000) at room temperature, the signal was detected using an ECL kit (Amersham) and visualized using Autoradiography.
Protein production
For protein production, HEK293T cells were transfected at 70% confluency in 6 × 10 cm plates. Cells were collected 48 h post-transfection and lysed using NP-40 lysis buffer (25 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 5% glycerol) supplemented with protease cocktail inhibitor, and the protein-containing supernatant was collected. Spike protein was purified from the supernatant via affinity purification column and quantified using BCA assay and confirmed with western blot. Produced spike protein was stored at -20 °C in 50% glycerol and thawed upon usage.
Cell cytotoxicity using MTT assay
MTT assay was performed on Vero cells to evaluate the cytotoxicity of the selected compounds as previously described (Loutfy et al. 2022; Mosmann 1983). Briefly, 104 cells per well were seeded in 96-well plates in DMEM complete media. The following day, cells were treated with different concentrations of the compounds in triplicates. After 48 h, MTT dye was added to the cells (0.5 mg/mL) and incubated for 2 h. The plate was then decanted and 100 µL of DMSO was added per well. Absorbance readings were obtained at 570 nm using a microplate reader (CLARIOstar plus, BMG LabTech, Germany). The cell viability was then expressed as the percentage of the untreated control and CC50 (defined as the concentration that reduced cell viability by 50%) values of the tested compounds were calculated by GraphPad Prism 8 software.
Validation of spike protein binding activity
To validate the binding activity of the produced spike protein (SP) versus the commercial spike protein (SC) we allowed it to interact with ACE2 using our previously published protein-protein interaction detection method (Emam et al. 2022). Briefly, small glass slides of equal size, 1 cm x 1 cm x 1 mm, were cleaned, coated with APTES, and then activated with EDC-NHS. ACE2 protein was immobilized on the slides, 100 nM on each slide, then they were rinsed with PBS. Both SP and SC were diluted to 2 concentrations 10 nM and 100 nM. Each slide was then incubated with one of the spike proteins followed by the anti-spike antibody (10 nM), then the FITC-labelled secondary antibody (10 nM) with rinsing between each incubation. Two negative controls were included in each run to test for specificity. The negative control was subjected to all the mentioned steps excluding the addition of spike protein.
Fluorescence intensity readings were recorded before and after adding spike protein and after the final wash by transferring slides into 24-well plates and using a microplate reader (CLARIOstar plus, BMG LabTech, Germany) for signal detection.
Standardization of the in-house immunofluorescent assay for drug screening
Chitosan nanoparticles were used as a drug for the optimization of the assay conditions. CNPs preparation and characterization were previously described (Loutfy et al. 2022). The modified assay was performed following the same procedures as described in the previous section except for an extra incubation step between the immobilized protein (ACE2) and CNPs for 40 min, followed by rinsing with PBS. In the current assay, various concentrations of CNPs (1, 5, 10, 20, 30, and 50 µg/ml) were used (Emam et al. 2021).
Following standardization, we screened the selected compounds: Curcumin (Cur), Gallic acid (GA), Quercetin (Q), and Silymarin (Sil). Taking into consideration the CC50 values of each compound, different concentrations ranging from 5 µg/mL to 500 µg/mL were used. The IC50 of each compound, which represented 50% inhibition of the substrate (ACE2) binding to its ligand (or to the spike protein), was determined using GraphPad Prism 8 software.
In each run, positive and negative controls were performed. However, the controls were counterintuitive as the negative control included the addition of spike & ACE2 proteins and the antibodies, producing a high signal. While for the positive control, we excluded the addition of spike protein, subsequently, producing no signal.


Comments (0)