
Remember me
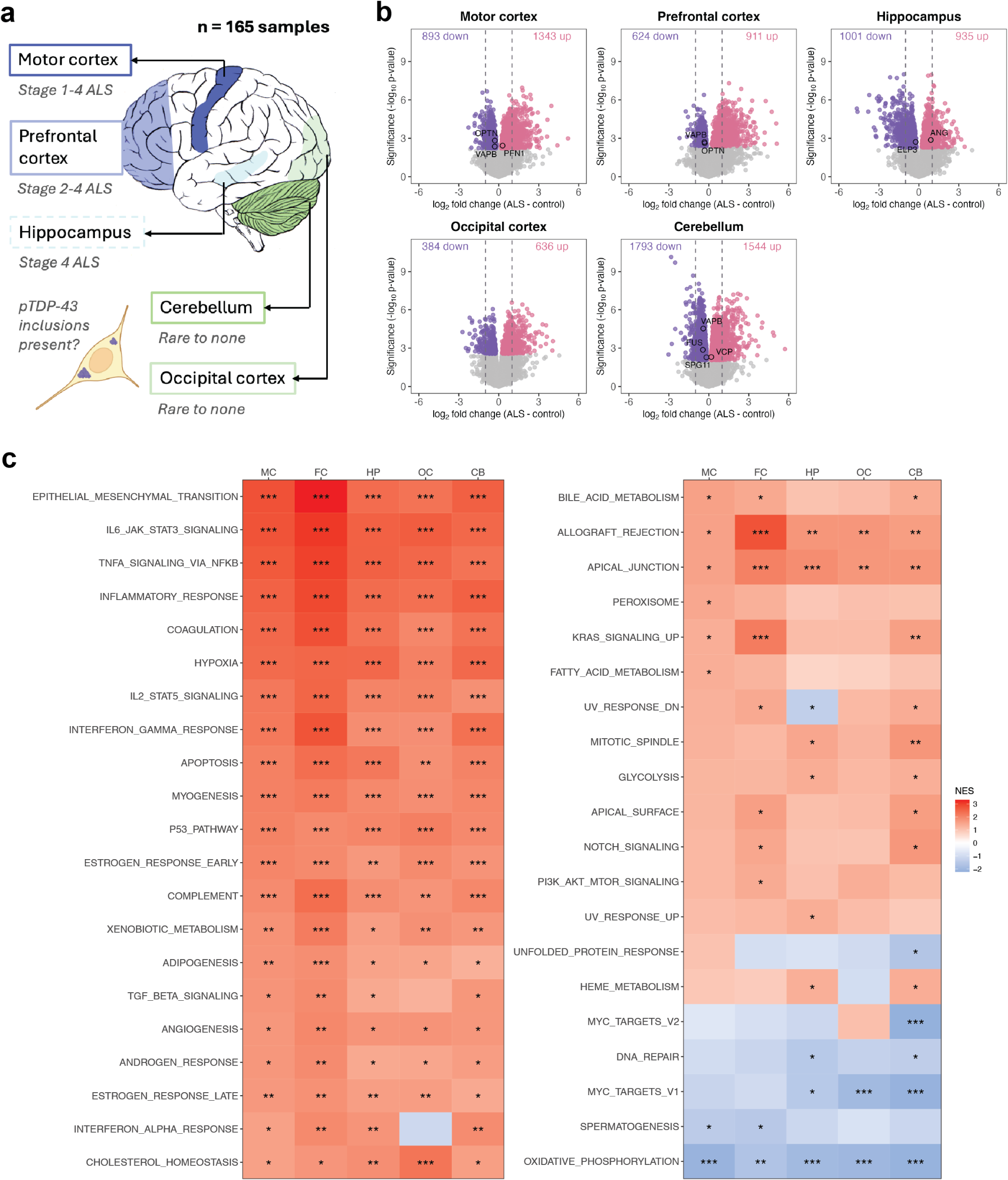





Aberrant accumulation of the RNA binding protein TDP-43 in the cytoplasm and its depletion from the nucleus are pathological hallmarks of several neurodegenerative diseases including subsets of frontotemporal lobar degeneration (FTLD-TDP), amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease (AD-TDP). Loss of TDP-43 from the nucleus impairs its ability to repress cryptic exon (CE) inclusion during RNA splicing [1]. Consequently, CEs are anomalously included in critical transcripts such as STMN2 and UNC13A [2,3,4,5,6,7,8,9], which can produce truncated or destabilized RNAs and lead to a loss of their function. Recently, we and others demonstrated that some transcripts with in-frame CEs produce stable CE-containing novel proteins detectable in cerebrospinal fluid (CSF) from patients with FTLD-TDP or ALS [10, 11]. One notable example is a cryptic protein derived from the gene hepatoma-derived growth factor-like protein 2 (HDGFL2-CE), a histone-binding protein expressed throughout the brain. Accordingly, CSF HDGFL2-CE could potentially serve as a sensitive biomarker of TDP-43 pathology, illuminating the contribution of pathological TDP-43 to the clinical variability in TDP-43 proteinopathies [11]. But testing whether CSF HDGFL2-CE is indicative of TDP-43 pathology is hampered by the lack of robust methods to measure pathological TDP-43 in biofluids. To determine if HDGFL2-CE abundance can be used as a readout of the presence of TDP-43 pathology, we probed whether HDGFL2-CE is preferentially expressed in affected neuroanatomical regions with TDP-43 proteinopathy in a cohort of well-characterized post-mortem tissues from FTLD-TDP and AD-TDP cases, and whether HDGFL2-CE abundance associates with pathological TDP-43 burden.
Based on the predicted structure of HDGFL2-CE protein (Fig. S1A), we had generated a previously described rabbit polyclonal HDGFL2-CE antibody (Mayo-LP) [10] that specifically detects HDGFL2-CE but not wild-type HDGFL2 (HDGFL2-WT) proteins in lysates from TDP-43-depleted human induced pluripotent stem cells (iPSC) (Fig. S1B). Using a commercial C-terminal HDGFL2-WT antibody as the capture antibody, and our Mayo-LP HDGFL2-CE antibody as the detection antibody, we then developed a Meso Scale Discovery (MSD) immunoassay that dose-dependently detected endogenous HDGFL2-CE protein in 500‒8000 ng of total protein lysate from TDP-43-depleted iPSC-derived neurons [10]. We have since further optimized the assay by biotinylating the capture antibody, using streptavidin MSD plates, and testing different diluents (Fig. S1C−E). When using MSD Diluent 35, our modified assay detected HDGFL2-CE but not HDGFL2-WT in 16 ng of total protein in lysates from HEK293T cells overexpressing these proteins (Fig. S1D). To determine whether our assay is sufficiently sensitive to detect endogenous HDGFL2-CE, we used lysates from control and TDP-43-depleted iPSCs. Compared to Diluent 35, Diluent 100 provided a better signal to noise ratio detecting endogenous HDGFL2-CE in as little as 125 ng of total protein from TDP-43-depleted iPSC lysates (Fig. S1E).
Next, we tested if our optimized assay could detect HDGFL2-CE in brain regions of FTLD-TDP (amygdala and frontal cortex) and AD-TDP (amygdala) characterized by TDP-43 pathology [7]. Compared to cognitively normal controls (controls), HDGFL2-CE was significantly increased in the amygdala of FTLD-TDP and AD-TDP cases in unadjusted analysis and when adjusting for age at death and sex (Fig. 1A, Table S1). In contrast, frontal cortex HDGFL2-CE was significantly increased only in FTLD-TDP cases when compared to controls (Fig. 1B, Table S1). When comparing AD cases without TDP-43 pathology (AD no TDP) to AD-TDP cases, HDGFL2-CE was significantly increased in the amygdala, but not the frontal cortex, in AD-TDP (Fig. 1A, Table S1) – an expected result given the paucity of TDP-43 pathology in the frontal cortex of AD-TDP cases.
Fig. 1
HDGFL2-CE proteins are increased in brain regions with TDP-43 pathology in FTLD-TDP and AD-TDP, and distinguish these TDP-43 proteinopathies from non-TDP-43 controls. Immunoassay quantification of HDGFL2-CE proteins in the amygdala (A) and frontal cortex (B) of cognitively normal controls (Ctrl, n = 27, n = 26 amygdala and n = 25 frontal cortex), FTLD-TDP (n = 67), AD-TDP (n = 70) and AD no TDP (n = 27). Data are presented as mean ± s.e.m. * P < 0.05 and **** P < 0.0001, ns: not significant. (C, D) Area under the receiver operating characteristic curves (AUC) showing the discriminatory capability of HDGFL2-CE in the amygdala or frontal cortex to distinguish FTLD-TDP from Ctrl (pink), AD-TDP from Ctrl (gold), and AD-TDP from AD no TDP (black). AUC values are shown. (E–G) Scatterplots of HDGFL2-CE protein and RNA abundance with pTDP-43 abundance in the amygdala (E) and frontal cortex (F) of FTLD-TDP patients, as well as in the amygdala of AD-TDP patients (G). Regression coefficients (β) and P values from linear regression analysis of pTDP-43 with HDGFL2-CE protein and RNA adjusting for age and sex are shown
The presence of HDGFL2-CE differentiated individuals with and without TDP-43 pathology in the amygdala: HDGFL2-CE distinguished between controls and individuals with TDP-43 pathology with an area under the receiver operating characteristic curve (AUC) of 0.85 and 0.92 for AD-TDP and FTLD-TDP, respectively, indicating good to excellent discriminatory ability (Fig. 1C). When assessing whether amygdala HDGFL2-CE protein distinguishes AD no TDP from AD-TDP, we found moderate discriminatory ability (AUC of 0.68, Fig. 1C). In the frontal cortex, HDGFL2-CE differentiated controls and FTLD-TDP with an AUC of 0.82, indicating good discriminatory ability (Fig. 1D).
Finally, phosphorylated TDP-43 (pTDP-43) burden in the amygdala and frontal cortex significantly associated with HDGFL2-CE protein and HDGFL2-CE RNA abundance in FTLD-TDP in both unadjusted analysis and in analysis adjusting for age at death, sex, and RNA integrity number (RIN), the latter for analysis of HDGFL2-CE RNA only (Fig. 1E, F, Table S2). In the amygdala of AD-TDP cases, pTDP-43 burden also associated with HDGFL2-CE protein and RNA in unadjusted and adjusted analyses (Fig. 1G, Table S2). However, estimated β coefficients where higher for HDGFL2-CE protein than HDGFL2-CE RNA indicating that HDGFL2-CE protein serves as a more accurate indicator of TDP-43 dysfunction.
Retention of CEs in mRNAs owing to TDP-43 dysfunction is well-documented in FTLD-TDP, ALS and AD-TDP [2,3,4,5,6,7,8], but the identification that certain in-frame CEs generate stable cryptic proteins is new [8, 10, 11]. Here, we explored the recently identified HDGFL2-CE protein. By developing a sensitive and specific immunoassay to detect HDGFL2-CE proteins, we observed that HDGFL2-CE is significantly increased in brain regions with TDP-43 pathology in FTLD-TDP and AD-TDP, associates with pTDP-43 burden, and can distinguish individuals with TDP-43 pathology from those without. In line with our findings, Irwin et al. used their HDGFL2-CE antibody to perform immunofluorescent staining of motor cortex and hippocampus tissues from patients with ALS-FTD demonstrating that HDGFL2-CE proteins accumulate in cells exhibiting pTDP-43 pathology [11]. They additionally found that, compared to controls, CSF HDGFL2-CE was statistically significantly higher in individuals likely to have TDP-43 pathology, namely presymptomatic or symptomatic C9orf72 repeat expansion carriers and patients with sporadic ALS [11].
Collectively, these findings show that the presence of HDGFL2-CE in the brain is a sensitive reporter of TDP-43 pathology in neurodegenerative diseases. These findings empower CSF HDGFL2-CE as a surrogate marker of TDP-43 pathology and dysfunction, which in turn would inform the selection of ideal participants for clinical trials of potential TDP-43-based therapeutics, and potentially enable precision medicine strategies for pathological subtypes of FTLD and AD.
Comments (0)