
Remember me
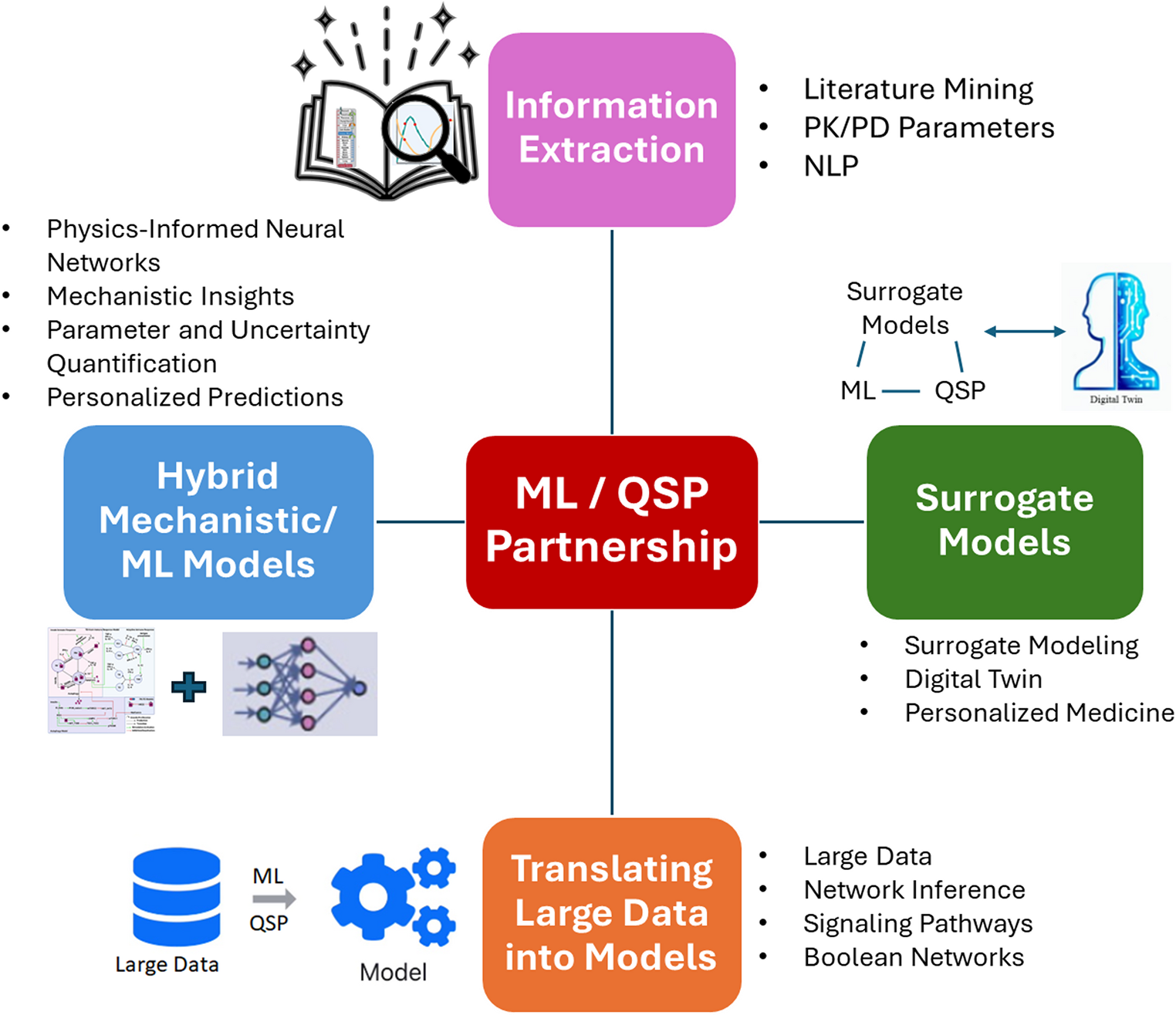



In this review, we would like to achieve the following objectives:
1).To provide a high-level perspective on the important role of MIDD in optimizing each stage of drug development, and to outline the frequent MIDD quantitative tools (Table 1), and to elucidate their general utilities addressing diverse relevant questions of interests.
2).To present a roadmap illustrating how commonly utilized PMx tools (Fig. 1) align with development milestones, guiding the progression from early discovery through regulatory approval, and ensuring that methodologies are appropriately matched to QOI in the COU.
Fig. 1
Illustration of commonly used MIDD tools across new drug discovery and development
3).To discuss the MIDD application in 505(b) (2), generic, and practice.
4).To list current challenges and opportunities.
In Table 1, we summarize some key tools that are frequently applied in MIDD.
Table 1 Commonly utilized MIDD toolsIn Fig. 1; Table 2, we illustrate and summarize the stages of drug development alongside relevant modeling tools. A common and practical question faced by development teams is: “Which models will provide the most useful insights for this indication at this stage?” The answer depends on selecting modeling strategies that are fit-for-purpose—aligned with the specific phase of development, intended indication, and the key scientific or regulatory questions being addressed.
For example, during early discovery and lead optimization, QSAR models, AI/ML tools for target prediction, or QSP frameworks may be most valuable for generating translational insights. In the preclinical stage, PBPK and semi-mechanistic PK/PD models, combined with allometric scaling, can be employed to project human pharmacokinetics and inform first-in-human (FIH) dose selection. During early clinical trials, Bayesian adaptive designs, population PK (PPK) models, and exposure–response (ER) models are particularly useful for characterizing variability and linking exposure to early safety or efficacy signals.
As development progresses, the modeling approach must evolve accordingly—from exploration to decision-making. PPK/ER models are frequently used for dose selection and justification in pivotal trials, while disease progression and model-based meta-analyses (MBMA) can support long-term efficacy projections or indirect comparisons. In the post-market phase, virtual population simulations and PBPK-based drug–drug interaction (DDI) models are often used to refine product labeling and meet post-approval commitments.
Given the complexity of drug development, along with organizational differences in strategy, resources, and timelines, we encourage developers to adopt fit-for-purpose MIDD practices—selecting and tailoring models that are most appropriate for their context. This approach ensures that modeling efforts are both efficient and impactful, supporting informed decisions and improving the likelihood of success across the product lifecycle.
(a) Models are listed in the alphabetical order; (b) including but limited to the models listed in Fig. 1. (c) FDA review various models across therapeutic areas, modalities and the unmet medical needs.
Table 2 New drug development phases and the corresponding modeling toolsDiscoveryDuring the discovery phase, MIDD leverages computational modeling and simulations to streamline the target identification and validation, as well as the lead compound optimization and nomination. By integrating multimodal data sources and predictive analytics, MIDD enables a more informed and strategic approach to discovering novel therapeutic candidates.
Target identification and validationMIDD uses advanced technologies like artificial intelligence (AI), machine learning (ML), multi-omics data, and bioinformatics to analyze multi-scale biological systems. AI and ML algorithms, building upon traditional quantitative structure-activity relationship (QSAR) models, can predict potential drug targets by analyzing large datasets to identify patterns and correlations that conventional methods might overlook [17,18,19]. Omics technologies, including genomics, proteomics, and metabolomics, provide deep insights into biological pathways and disease mechanisms, enabling more precise and effective target selection through computational modeling [20, 21]. QSP, with their mechanism-based nature, can also be helpful in target validation.
Lead compound optimization and nominationMIDD uses AI/ML models to predict the preclinical ADME properties of potential compounds. These models allow for rapid screening of extensive chemical libraries, significantly reducing time and resources needed for experimental testing. Enhanced QSAR models integrated with AI/ML techniques offer better predictive accuracy by considering a broader range of molecular descriptors and biological interactions. This accelerates the optimization of lead compounds, enabling early identification of candidates with optimal therapeutic profiles.
As real-world examples, the collaboration between Insitro and Bristol-Myers Squibb (BMS), showcased how ML-driven drug discovery promises to disentangle the complexity of disease (e.g., amyotrophic lateral sclerosis, ALS), and identify novel genetic targets, to potentially discover disease-modifying medicine [22]. The proprietary platform, Insitro Human (ISH), combines induced pluripotent stem cell derived disease modeling, ML analysis, human genetics and genomics to support in vitro models, hence promising to identify disease progression and patient segments, and discover potential targets [23]. Similarly, ML has been used to determine new ALS-associated genes, potential targets, critical biomarker pathways, and their associations with the disease subtypes and tissue samples [24, 25]. As another example, AstraZeneca’s collaborations with numerous external digital partners demonstrate how MIDD strategies could enhance drug discovery pipelines and highlighted the growing industry-wide adoption of these technologies [26]. Case studies of some of the most demanded drug products further illustrate the benefits of MIDD in drug discovery. For example, in the development of Semaglutide, structure modeling of the receptors provided essential insights into its ligand recognition and activation [27, 28]. Specifically, the authors reported the high-resolution crystal structure with molecular details between the ligand and the receptor, revealing major confirmational changes in secondary structure during the binding and key interactions with the peptide ligand. They further identified a dual-binding trigger model [28]. The development of EGFR (Epidermal Growth Factor Receptor) inhibitors also demonstrated the power of theoretical analysis and modeling [29, 30]. Notably, guided by the QSAR modeling along with a glutathione-based assay, a series of EGFR inhibitors were designed and synthesized to target a cystine residue in the ATP binding site, showing improved activity to overcome the mutations by gefitinib and erlotinib [30]. These model-informed insights facilitated the design and optimization of effective inhibitors, predicting their binding affinities and immunomodulatory effects, which accelerated their progression from discovery to clinical application.
Preclinical researchThe Preclinical Research phase focuses on assessing ADME properties of a drug, and preclinical toxicology assessment. PBPK modeling is an advanced tool used to simulate and predict the ADME properties of a drug in humans. By integrating detailed physiological, biochemical, and drug-specific data, PBPK models help researchers understand a drug’s distribution and elimination processes, which is invaluable for translating preclinical findings into human contexts. In addition to PBPK modeling, semi-mechanistic PK/PD modeling plays a vital role in drug development. This approach combines empirical data with mechanistic insights, providing a nuanced understanding of the relationship between drug concentration and its pharmacological effects. Semi-mechanistic PK/PD models, including both efficacy and toxicity, offer a flexible framework that adapts to varying levels of biological complexity, enhancing predictions of human PK and optimizing dose selection.
A primary application of these modeling techniques is projecting human pharmacokinetics from preclinical data. By leveraging PBPK and semi-mechanistic PK/PD models, researchers can predict how a drug is likely to behave in humans, including its absorption, distribution, metabolism, and elimination profiles. This information is essential for determining the starting dose for FIH trials [31, 32]. Accurate dose projections reduce the risk of adverse effects while ensuring therapeutic levels of the drug are achieved in the human body [33]. Ultimately, the goal of ADME assessment, PBPK modeling, and semi-mechanistic PK/PD modeling is to translate preclinical findings into a safe and efficacious dose for humans. By integrating data from various modeling approaches, researchers can identify a dose that is both safe and effective for the target population, thereby ensuring that new therapies provide real-world benefits to patients.
QSP models play a significant role in preclinical research by integrating biological, pharmacological, and mathematical principles to predict drug effects within biological systems. By leveraging QSP models in preclinical drug development, researchers can better optimize dosing regimens, discover reliable biomarkers, and predict efficacy and toxicity profiles, significantly reducing clinical trial failures and enhancing informed study designs throughout the pharmaceutical development process.
With their high potential, QSP modeling has been increasingly applied in the pharmaceutical industry [34]. For the research of CNS diseases, QSP modeling has been recognized as an powerful extension of the more traditional modeling methods [35]. Additionally, QSP models have been developed to evaluate the cardiovascular safety drugs was and enable risk mitigation [36].
Clinical researchFIH studies represent a major milestone in drug development, marking the first time a new drug is investigated in humans. These studies assess the safety, PK, and possibly exploratory efficacy and PD of a drug, in healthy volunteers or patients. In recent years, (QSP) modeling has gained prominence in FIH studies. By offering a comprehensive understanding of the intricate interactions between in vitro cellular data, preclinical toxicity and PK, the underlying disease mechanisms, and the drug properties, QSP may support the selection of substantially higher starting doses for FIH trials compared to traditional methods like minimum anticipated biological effect level (MABEL). The predicted starting doses were 5 mg/kg and 0.045 to 0.1 mg/kg, using QSP and MABLE methods, respectively, with 50–100 folds higher [37].
This approach was accepted by FDA and Australian Human Research Ethics Committee, and accelerated the dose escalation of FIH trial, reducing the sub-efficacious doses to patients and saving valuable time and cost. Additionally, modified MABEL approach was proposed, using the most relevant rather than the most sensitive measure of pharmacological activity [38, 39]. Specifically, the median EC50 from the primary cell killing, rather than using EC10-30% in the most sensitive cell assay, improving the starting dose to be closer to the efficacious dose to patients [38]. By integrating biological, disease-specific data, and drug pharmacological data, QSP models also promise to enhance the ability to predict drug responses, optimize dosing strategies, and reduce the risk of adverse events during early trials.
In early-phase clinical development, a common challenge arises: “We have very limited patient data in FIH trial—how can we accelerate via MIDD?” Quite often, it would benefit from maximizing prior knowledge. Baysesian model-based adaptive design is increasingly applied, aiming to reduce the sample size in human trials, by leveraging data derived from in vitro, preclinical or human data. integrating in vitro pharmacology, preclinical PKPD, and early clinical human data (ref: current reference list 39). Additionally, mechanistic models such as PBPK and QSP, along with virtual population simulation, would derisk the uncertainties [40,41,42]. With increased number of participants, PPK plays a more vital role in FIH studies and beyond, describing the typical pk parameters and quantifying the variability in drug concentrations across individuals. PPK modeling helps to identify covariates such as age, weight, renal function, or genetic factors that affect the drug exposure measures. More importantly, drug exposures with high variability and/or non-linear pk would necessitate the exposure-safety analysis than the conventional dose-limiting toxicity approach. Quantifying the variability or uncertainties further allows for optimizing dose and dosing regimens with FIH trials, where the enrolled patients are “all comers”, in oncology therapeutic area, thereby enhancing drug benefit risk ratios. For example, exposure-toxicity guided Bayesian design vs. the dose-toxicity design was postulated in a Phase I dose escalation trial, with 15 patients administered with four different dose levels. The work demonstrated that exposure-based models would improve the selection of optimal dose, when the drug exhibits non-linear pk or large inter-subject variability [43]. Additionally, PPK modeling helped to determine appropriate dosing in different age groups and special populations, ensuring broad and safe vaccine deployment [44].
Exposure-Response (ER) analysis is another indispensable aspect in drug development. It involves characterizing the relationship between various drug exposures (and their efficacy or safety endpoints. ER models are essential in clinical studies to recommend RP2D dose, prior to the pivotal trial. For instance, Stelara (ustekinumab), used to treat autoimmune diseases like psoriasis and Crohn’s disease, relies on ER modeling to balance efficacy against potential side effects. For Stelara, ER analysis determined the dose that maximizes therapeutic benefits while minimizing risks [45, 46]. After the pivotal clinical trial, the PPK and ER models have been frequently applied for dose confirmation and benefit/risk assessment, along with clinical efficacy and safety data.
The integration of QSP, PPK, and ER models throughout drug development creates a robust framework for making well-informed decisions. These models allow decision makers to simulate various scenarios, predict outcomes in diverse patient populations, and support regulatory submissions. As discussed, the combination of PPK and ER models during the development of Comirnaty facilitated the rapid optimization of dosing strategies, which was critical under the accelerated timelines of the COVID-19 pandemic. Similarly, QSP models for Stelara provided a deeper understanding of its mechanism of action, enabling more refined clinical use across different inflammatory conditions. Major health authorities encourage the sponsors to discuss these MIDD approaches with the agencies in early clinical development. They have issued many draft or final guidance, published papers, hosted MIDD-centric workshops, designated regulatory pilot programs, or formed modeling & simulation working group [47,48,49,50].
In summary, FIH and the pivotal clinical trials, together with QSP, PPK, and ER models, are revolutionizing drug development by offering a precise and predictive understanding of drug behavior in humans. These models not only help the drug developers to maximize the benefit risk ratios of new therapies but also advance personalized medicine approaches. Success stories like Comirnaty and Stelara underscore the essential role these models play in bringing life-saving therapies to market and enhancing patient outcomes worldwide.
FDA reviewApplications of MIDD principles in FDA reviews started in early 1990 s and have continuously evolved to the transformative presence today [5]. The scope of MIDD applications is across all therapeutic areas, across adults and pediatric populations, and across stages of drug development such as EOPI/II meetings and the label update. With the emergence of advanced science and technologies, the methodologies have expanded from the conventional PBPK-based to further mechanistic QSP modeling, from PPK/ER based models to early adoption of AI/ML models, and from earlier applications to the standardized plan, data, analysis and reporting to regulatory bodies. Dosage selection, optimization, recommendation and confirmation, has been implemented in non-oncology disease therapeutic areas, via the totality of evidence of relevant preclinical data, doing ranging studies of clinical efficacy and safety, pharmacometrics analyses, and pivotal clinical trials. In recent years, dosage optimization becomes a mandate in oncology drug development and regulatory approval [51]. It aims to maximize the ratio between effective and safe dosage for patients, ensuring that the drug achieves its intended therapeutic effects while minimizing adverse events. The historic maximum tolerant dose (MTD) approach is more suitable for cytotoxic agents than the target or immunological therapies. Additionally, low-grade yet long-term toxicities may compromise the patient’s life of quality and reduce patient tolerability. Based on a recent IQ survey, “getting the dosage right” is a case-by-case approach, advocating the importance of multi-function collaborations with the focus on patient-centered paradigm [52]. The field has evolved significantly with strategic framework, deeper understanding of cancer biology, emerging treatment modalities, inherently diverse nature of tumor sub-types, therefore the non-oncology dose optimization paradigm may necessitate modifications for mostly life-threatening oncology therapeutic areas. The totality of dose optimization largely is composed of the utilization of clinical dose/exposure with the efficacy and safety is essential and quite often available for submission, while PD data at site of action is rarely available for dose selection or optimization [52]. Translational biomarkers, such as longitudinal tumor growth dynamics across clinical visits, and IL-6 release as a CRS surrogate for T-cell engagers, have provided supporting evidence for dose optimization [53, 54].
The recent methodologies include the integration of advanced tools like ML is increasingly valuable in dose optimization. ML models can analyze vast amounts of data from clinical trials, real-world evidence, and other sources to identify patterns that may not be apparent through traditional analysis. For example, during the COVID-19 pandemic, ML models helped optimize dosing regimens for various treatments, ensuring that patients received the most effective and safe doses for antiviral drugs and monoclonal antibodies [55, 56]. Additionally, AI/ML approaches, including both elastic net regression and artificial neural network, were independently applied to predict whether a patient is to benefit from Anakinra treatment, using the score rule of soluble urokinase plasminogen activator (suPAR) value of 6 ng/mL based on the SAVEMORE trial data. With 30 variables available from the baseline characteristics of the trial patient’s data, elastic new regression was to select the contributing features, and the neural network model was independently to rank the features and the cut-off values. Both methods resulted in consistent eight criteria for patient suPAR prediction. Further, the model was externally validated using SAVE trial [55, 56]. The application of ML, an emerging MIDD tool, offers a powerful predictivity of patient selection with benefits of the treatment. This AI/ML approach is especially valuable in therapeutic areas where patient responses can vary widely, especially during a global health crisis [57].
The future of MIDD, including dose optimization, lies in further integrating and refining the existing and emerging tools. As more clinical data becomes available, the role of ML and AI in dose optimization will expand. It is just the beginning, demonstrating how advanced modeling techniques and innovative technologies can lead to safer, more effective therapies for patients.
Post-market stageMIDD continues even after the market authorization, and the earlier applications are the post-marketing commitment/requirement implemented for dose optimization for several approved drug products, such as ponatinib [58] and ceritinib [59].
Different types of modeling play diverse role in post-market stage by enabling the continuous evaluation of a drug’s safety, efficacy, and real-world effectiveness after the initial regulatory approval. Pharmacometric models, such as PPK PKPD, and ER models, help refine dose adjustments for specific patient subgroups based on real-world variability. ML and AI-driven models analyze vast post-marketing surveillance data, including electronic health records (EHRs) and spontaneous adverse event reports, to detect rare but serious safety signals more efficiently than traditional methods. Real-world evidence (RWE) models, incorporating data from observational studies and registries, assessing long-term treatment outcomes, adherence patterns, and comparative effectiveness in diverse populations. Systems pharmacology and mechanistic models contribute by predicting potential off-target effects or long-term biological consequences based on known drug interactions. By integrating these diverse modeling approaches, post-market studies can enhance drug safety monitoring, optimize therapeutic use, and support regulatory submission in response to emerging risks or new clinical evidence.
Recent cases include but not limited to formulation change, dosage adjustment and label update. Published examples are nivolumab (often referred to as “Nivo”) and Pembrolizumab (“Pembro”) are both immune checkpoint inhibitors used primarily to treat cancers such as melanoma, non-small cell lung cancer (NSCLC), and renal cell carcinoma. These drugs work by targeting and blocking the programmed death-1 (PD-1) receptor on T-cells. Normally, the PD-1 receptor interacts with its ligand, PD-L1, often expressed by cancer cells, leading to the suppression of the immune response. By blocking this interaction, these drugs release the “brakes” on the immune system, allowing it to recognize and attack cancer cells more effectively. This ability to enhance the immune response against cancer has made them critical components of modern oncology treatments. One example is that pembrolizumab has been particularly notable for introducing a dosing schedule of every six weeks (Q6W) instead of the more frequent every three-week interval [60, 61]. This extended dosing schedule offers several advantages, including increased convenience for patients, reduced healthcare burdens, and potentially improved patient adherence to treatment. This regime is particularly advantageous for patients requiring long-term treatment, reducing hospital visits while maintaining efficacy and safety.
In some cases, Nivolumab or Pembrolizumab may be used in combination with other types of treatments in clinical trials. Combining these immune checkpoint inhibitors can enhance anti-tumor responses, but it may also increase the risk of immune-related adverse events. The decision to use these drugs in combination depends on the specific type of cancer, the patient’s overall health, and underlying conditions that could increase side effects. Clinical trials continue to explore these combinations’ safety and efficacy for different cancers [62, 63].
Prevnar, a pneumococcal conjugate vaccine, provides protection against Streptococcus pneumoniae, the bacterium responsible for infections like pneumonia, meningitis, and sepsis [64, 65]. When patients are receiving immunotherapy such as Nivolumab or Pembrolizumab, vaccination strategies must be carefully considered due to the immune system modulation these treatments cause. Oncologists often work closely with healthcare providers to ensure cancer patients are appropriately vaccinated, considering the timing and type of vaccines to optimize cancer treatment while maintaining overall health [64, 65].
Comments (0)