
Remember me


All constructs in this study were generated using standard molecular biology techniques. Mutant constructs were generated by QuikChange Site-Directed Mutagenesis, using the oligonucleotides listed in Supplementary Table 1. Construct sequences were verified through DNA sequencing (Microsynth Seqlab). Constructs for insect cell expression were cloned into pLIB vectors61 to allow generation of bacmids in DH10EmBacY62. Subsequently, baculoviruses were generated in Sf9 insect cells (Thermo Fisher Scientific cat. no. B85502) and were further used to infect Hi5 insect cells (BTI-TN-5B1-4; Thermo Fisher Scientific, cat. no. 11496016) for expression.
Bacterial expression of Ub, Ub variants, E2 UBE2L3, and DUBs OTUB1* and TRABID was performed by transforming corresponding plasmids into E. coli BL21(DE3) Rosetta cells. Single colonies were picked to inoculate precultures in LB medium supplemented with appropriate antibiotics, which were further diluted 1:100 into TB medium containing antibiotics. Cells were grown at 37 °C and 180 rpm to an optical density of 0.8; then, the temperature was lowered to 16 °C, and expression was induced by addition of 0.5 mM isopropyl β-d-thiogalactopyranoside (IPTG). After 18 h, cells were collected by centrifugation at 7,278g and 4 °C for 15 min.
Pellets were resuspended in ice-cold lysis buffer (50 mM Tris-HCl, pH 8, 200 mM NaCl, 5 mM DTT (or β-mercaptoethanol for histidine-tagged constructs) and 2.5 mM PMSF, and additionally 10 μg ml−1 leupeptin, 20 μg ml−1 aprotinin and 10 μg ml−1 DNAse I for insect cells). Cells were lysed through sonication followed by centrifugation at 50,000g for 30 min to separate cell debris.
C-terminally hexahistidine-tagged Ub constructs (Ub–6×His) were purified as described previously9. In brief, lysates were subjected to Ni-NTA affinity chromatography, followed by size-exclusion chromatography in 25 mM HEPES, pH 7.5, and 150 mM NaCl.
GST–human rhinovirus 3C protease cleavage site (3C)–Ub variants (GST–3C–Cys–UbK6R K11R K27R K29R K33R K48R K63R for fluorescent labeling, and GST–3C–UbK29C for the K29-linked chain formation probe) were pulled down through GSH affinity chromatography, followed by cleavage with 3C protease, leaving a residual GP dipeptide at the N-terminus (or GPC for Cys–UbK6R K11R K27R K29R K33R K48R K63R (Ub K0)). Cleaved Ubs were further purified by size-exclusion chromatography in 25 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM DTT. The former variant, when fluorescently labeled as described below, is referred to as *Ub(K0) in the main text and is used in pulse–chase ubiquitylation assays.
Untagged Ub and variants were purified through acetic acid precipitation. Lysates were acidified by the gradual addition of acetic acid; they were then stirred at room temperature until the pH reached ~4.5 and centrifuged at 20,000 rpm for 30 min. The supernatant was dialyzed against 25 mM sodium acetate, pH 4.5, at 4 °C overnight, and was then centrifuged again at 20,000 rpm for 30 min to clear any further aggregates. The supernatant was subjected to cation exchange chromatography followed by size-exclusion chromatography in 25 mM HEPES, pH 7.5, and 150 mM NaCl.
The E2 UBE2L3 was expressed as a N-terminal GST fusion with a 3C protease cleavage site and purified by GSH affinity chromatography, followed by on-bead cleavage with 3C protease overnight. The cleaved protein eluate was then subjected to anion exchange chromatography, during which UBE2L3 was collected in the flowthrough, and size exclusion chromatography in 20 mM Tris, pH 7.5, 150 mM NaCl, and 1 mM DTT.
OTUB1* was purified as described previously16, using Ni-NTA affinity chromatography followed by size exclusion chromatography in 25 mM HEPES, pH 7.5, 150 mM NaCl, and 2 mM DTT.
The TRABID catalytic domain (residues 245–697) was expressed as an N-terminal GST fusion with an intervening tobacco etch virus (TEV) protease cleavage site and purified through GSH affinity chromatography. Bound protein was eluted and further purified by anion exchange chromatography, followed by size-exclusion chromatography in 25 mM HEPES, pH 7.5, 150 mM NaCl, and 2 mM DTT.
The E1 UBA1 was expressed in insect cells as an N-terminal GST fusion with a TEV protease cleavage site and isolated by GSH affinity chromatography, followed by TEV protease cleavage overnight. Cleaved protein was further purified by anion exchange chromatography and subsequent size-exclusion chromatography in 25 mM HEPES pH 7.5, 150 mM NaCl, and 1 mM DTT.
TRIP12 constructs were expressed in insect cells as N-terminal GST fusions with a TEV protease cleavage site. For full-length TRIP12 (and corresponding point mutants), additional NaCl was added to resuspended pellets to a final concentration of 1 M before sonication. Protein was purified using GSH affinity chromatography and cleaved on beads by incubation with TEV protease overnight. Cleaved protein was washed off the beads and subjected to anion exchange chromatography, followed by size-exclusion chromatography in 25 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM TCEP. The TRIP12478–2040 variant (termed TRIP12ΔN, lacking the N-terminal intrinsically disordered region) was purified in a similar manner, but NaCl was not added before lysis and cation exchange chromatography was used.
WT UBR5 was prepared as described previously9. In brief, baculoviruses with a TwinStrep-GFP-UBR5 construct, generated in Sf9 cells, were used for expression in HEK293S cells (CRL-3022, ATCC). UBR5 was purified through Strep affinity chromatography, overnight cleavage using 3C protease, and subsequent size-exclusion chromatography in 25 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM TCEP.
Synthetic Ubs containing lysine or lysine analogs with different side chain lengths (one methylene (Dap), l-2,3-diaminopropionic acid; two methylenes (Dab), l-2,4-diaminobutyric acid; three methylenes (Orn), l-ornithine; or five methylenes (hLys), l-homolysine) in position 29, and an additional aspartate at the C-terminus for use in di-Ub generation (see below), were synthesized through solid-phase peptide synthesis, as described in Supplementary Note 1.
Generation of Ub chains with different linkage typesK27-, K29-, and K33-linked di-Ubs were synthesized as reported previously9. M1-linked di-Ub was produced recombinantly as a linear fusion with an N-terminal GST-tag in E. coli and purified as described previously9.
K6-, K11-, K48-, and K63-linked Ub chains were prepared as follows, using enzymatic assembly with recombinantly produced tagless Ub.
K6-linked chains were generated by incubating 2.5 mM Ub with 0.1 μM E1, 0.6 μM UBE2L3, 10 μM NleL in 40 mM Tris-HCl, pH 8.8, 10 mM MgCl2, 1 mM DTT, and 10 mM ATP for 3 h at 37 °C. The reaction was quenched with 10 mM DTT, and by-product K48-linked Ub chains were removed by subsequent incubation with 2 μM OTUB1 for 3 h at 37 °C.
K11-linked Ub chains were generated by incubating 0.5 mM Ub with 0.25 μM E1 and 5 μM Ube2S-UBA-IsoT63 in the presence of 10 mM ATP for 2 h at 37 °C.
To generate native K48-linked Ub chains, 2.5 mM Ub was incubated with 1 μM E1 and 25 μM UBE2R1 in the presence of 10 mM ATP for 3 h at 37 °C. The reaction was quenched by adding 10 mM DTT and 1 μM associated molecule with the SH3 domain of STAM (AMSH).
K63-linked Ub chains were generated by incubating 1 mM Ub with 0.5 μM E1, 8 μM Ube2N, and 8 μM Ube2V1 in 40 mM Tris-HCl, pH 8.5, 10 mM MgCl2, 0.5 mM DTT, and 10 mM ATP at 37 °C for 30 min, and were then quenched by the addition of 10 mM DTT.
Different chain lengths of the various chain types were consecutively separated using several rounds of cation exchange chromatography, followed by size-exclusion chromatography in 25 mM HEPES pH 7.5, of which only di-Ubs were used in this study.
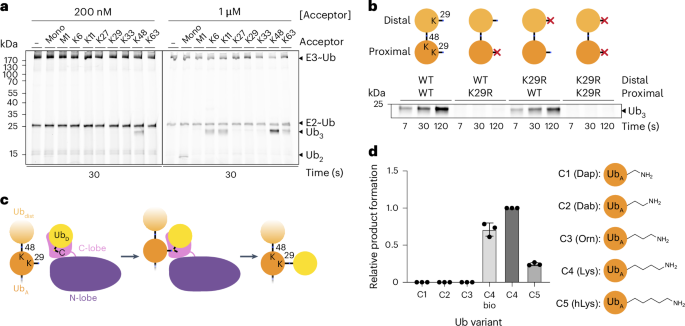
Generation of mutant K48-linked di-UbsSpecific mutant K48-linked di-Ubs were produced by incubating 275 µM donor Ub (carrying the K48R substitution to prevent longer chain formation, and optionally the K29R substitution as indicated) and 250 µM acceptor Ub (D77 variants with an additional Asp at the C-terminus to prevent usage as donor, along with other substitutions as indicated) with 20 µM UBE2R2 and 1 µM E1 in 25 mM HEPES pH 7.5, 150 mM NaCl, 2.5 mM MgCl2, 12.5 mM ATP and 1 mM DTT at 37 °C overnight. Products were isolated using cation-exchange chromatography and size-exclusion chromatography in 25 mM HEPES, pH 7.5, and 150 mM NaCl (also with 1 mM TCEP for K29C variant). These di-Ubs are referred to in following sections as UbK48R–UbD77, with the distal Ub listed first, followed by the proximal (and principal acceptor) Ub.
Fluorescent labelingTo generate fluorescently labeled donor Ub for pulse–chase assays, Cys–Ub K0 (with all lysines mutated to arginine to prevent chain formation) was purified as described above. Purified protein was preincubated for 15 min on ice with 1 mM DTT to fully reduce the N-terminal cysteine, followed by desalting twice using PD10 (GE Healthcare) desalting columns to remove reducing agents. Desalted Ub was then incubated with fivefold molar excess of fluorescein-5-maleimide (AnaSpec) at room temperature for 2 h. Products were again desalted twice and subjected to size-exclusion chromatography in 25 mM HEPES, pH 7.5, and 150 mM NaCl to remove excess dye.
Generation of stable complexes mimicking polyubiquitylation by TRIP12Specialized chemical-biology tools were used to covalently trap TRIP12 with its catalytic Cys, and donor and acceptor Ubs were stably linked to mimic the transition state in Ub transfer (Fig. 2a). Their basic building block, Ub–BmDPA (6×His–Ub1–75–(E)-3-(2-(bromomethyl)-1,3-dioxolan-2-yl)prop-2-en-1-amine), was synthesized as described previously9. In brief, 6×His–Ub1–75–intein–chitin binding domain (CBD) was expressed in E. coli and purified through Ni-NTA affinity chromatography. Next, the split intein fragment was thiolytically cleaved using sodium 2-mercaptoethane sulfonate (MESNa) to form 6×His–Ub1–75–MESNa, which was further purified using size-exclusion chromatography (25 mM MES, pH 6.2, and 100 mM NaCl). Then, 6×His–Ub1–75–MESNa (5 g L–1) was conjugated to BmDPA (ChiroBlock, 0.4 M) in the presence of 10 mM N-hydroxysuccinimide at 30 °C and 300 rpm overnight. The resulting Ub–BmDPA species was subsequently deprotected using 40 mM p-toluenesulfonic acid in 54% (vol/vol) trifluoroacetic acid. It was then precipitated and washed using cold diethyl ether, and refolded to yield Ub–ABPO (6×His–Ub–(E)-5-amino-1-bromopent-3-en-2-one) as the final product. Reactive probes were formed by prereducing 100 µM UbK29C (for K29-linked chain formation probe) or UbK48R–UbK29C D77 (for K29/K48-linked branched chain formation probe, synthesis described above) with 1 mM TCEP. The mixture was then desalted (Zeba Spin Desalting Column, 3 K MWCO) and combined with an excess of Ub–ABPO (optimized for each batch in small-scale test reactions), which was then incubated at 30 °C for 1 h. Products were isolated using Ni-NTA affinity chromatography followed by size-exclusion chromatography in 25 mM HEPES, pH 7.5, and 150 mM NaCl.
Chemically stable mimics of ubiquitylation complexes were prepared by incubating freshly purified TRIP12 (full-length or ΔN) at 2 µM with excess of reactive probe (10-fold for K29/K48-linked branched chain formation, 15-fold for K29-linked chain formation) in 25 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM TCEP at 30 °C for 1 h. Reactions were quenched by 1:1 dilution in 25 mM HEPES, pH 7.5, 150 mM NaCl, and 5 mM DTT, and were immediately plunge-frozen to prepare samples for cryo-EM.
Cryo-EMSample preparationCHAPSO was added to samples at a final concentration of 4 mM right before plunge-freezing. Four microliters of sample was applied on glow-discharged R1.2/1.3 200 mesh holey carbon grids (Quantifoil) and plunge-frozen in liquid ethane on a Vitrobot Mark IV (Thermo Fisher Scientific; blot force 3, blot time 3 s, 4 °C, 100% humidity).
Data collectionGrids were screened using SerialEM v4.1 on a Talos Arctica cryo-TEM (FEI), operated at 200 kV and equipped with a K3 direct electron detector (Gatan), to select grids suitable for high-resolution data collection. Screening datasets for TRIP12ΔN samples were collected at a magnification of ×22,000 and pixel size 1.841 Å px–1, in a defocus range of −1 to −2.6 µm, and with a dose of 60.0 e– per Å2 fractionated over 40 frames.
Final high-resolution datasets were collected using SerialEM v4.1 on a Titan Krios cryo-TEM (FEI) operated at 300 kV, equipped with a Bio Quantum post-column energy filter (Gatan; 10 eV) and K3 direct electron detector (Gatan) in counting mode, at a magnification of ×105,000 and pixel size 0.8512 Å px–1. Defocus was varied from –0.6 to −2.2 µm. Samples were exposed for 3 s. Average total exposure doses were 76.5 e– per Å2 for the full-length TRIP12 branched chain formation dataset, 64.8 e– per Å2 for the TRIP12ΔN branched chain formation dataset, and 66.8 e– per Å2 for the TRIP12ΔN chain formation dataset, fractionated over 30 frames. Detailed data collection, refinement and validation statistics are summarized in Table 1.
Data processingFor the dataset with full-length TRIP12 representing forging of a K29/K48-linked branched chain, dose weighting, motion correction and CTF estimation were performed in RELION 5.0 (ref. 64) using the built-in implementation of MotionCor2 and CTFFIND 4.1 (ref. 65), respectively. Particles were picked using Gautomatch (K. Zhang, MRC Laboratory of Molecular Biology) and extracted in RELION (4× binned, box size 80 px, applies to all datasets), then imported into cryoSPARC v4.5 (ref. 66) for further 2D and 3D classification, re-extraction at full box size (320 px for all datasets), and high-resolution refinement. A detailed processing schematic is provided in Extended Data Fig. 2. The data obtained with full-length TRIP12 exhibited substantial orientation bias (see also Extended Data Fig. 2c), resulting in anisotropy of the final reconstruction, especially at higher resolution, which hindered accurate building of atomic models. Use of TRIP12ΔN largely eliminated this orientation preference (Extended Data Figs. 4c and 5c and Supplementary Video 1).
Raw movies of all TRIP12ΔN datasets were processed in cryoSPARC v4.5, using Patch Motion correction followed by Patch CTF estimation. For screening datasets, particles were picked using blob picker and processed as displayed in Extended Data Fig. 3. Maps obtained from screening datasets were used as reference volumes for heterogeneous refinements in the high-resolution datasets. For the high-resolution TRIP12ΔN K29/K48-linked branched chain formation dataset, particles were picked using templates generated with the map obtained from the screening dataset, and classified and refined as depicted in Extended Data Fig. 4. Three-dimensional variability analysis (as shown in Supplementary Video 2) was performed on the particle set before 3D classification, filtering the resolution to 6 Å and solving for 6 modes. To highlight the similarities and variations in conformations in Supplementary Video 2, maps were fit with the following individual units extracted from the final refined coordinate file: (1) the TRIP12 ARM domain, (2) the TRIP12 HEL-UBL domain and HECT domain N-lobe, (3) the distal Ub, and (4) the TRIP12 HECT domain C-lobe, C-terminus, and acceptor and donor Ubs.
For the dataset representing TRIP12ΔN producing a K29-linked Ub chain, particles were picked using blob picker (subsequent template picking did not yield an improved particle set) and processed as outlined in Extended Data Fig. 5. Further 3D classification and 3D variability analysis steps, beyond those given in processing schemes, were explored, but did not yield an improved resolution or reconstruction.
Maps were sharpened using DeepEMhancer (version 2020.09.07)67. Initial models were built based on Alphafold2 multimer68 predictions of TRIP12 in complex with Ub (which was placed as donor Ub and around the distal Ub binding site, both in ‘loop-out’ conformation). Because the dataset representing TRIP12ΔN building a K29-linked chain produced the highest-resolution map, especially in the active site, this structure was used as a starting model for building and refinement of the structure representing branched chain formation. Because density for distal Ub was lower resolution and visualized at lower contour, this Ub was largely rigid-body docked in UCSF ChimeraX v1.8. Densities for the K48 linkage in the di-Ub acceptor and for the probe molecule in the active site were absent or visible only at a very low threshold in the deepEMhancer-sharpened map, but could be modeled on the basis of the unsharpened map. Initial models were refined using iterative cycles of manual fitting in Coot69 and real-space refinement in Phenix70. Structural renderings were created using UCSF ChimeraX v1.8.
Biochemical assaysPulse–chase assays were performed to track the transfer of fluorescently labeled donor Ub (*Ub(K0)) from E2 to E3 enzymes and further onto substrate. UBE2L3 was used as the E2 because a corresponding activity-based probe showed higher reactivity toward TRIP12 than did a UBE2D2-derived probe in a previous study71.
For experiments assaying activity of TRIP12, unless indicated otherwise, 7.5 µM fluorescently labeled Ub K0 (henceforth referred to as *Ub(K0)) and 6 µM UBE2L3 were incubated with 0.3 µM UBA1 in 50 mM HEPES, pH 7.5, 100 mM NaCl, 2.5 mM MgCl2, 1.5 mM ATP, and BSA (0.05 g L−1) at room temperature for 30 min. The pulse reaction was quenched by 1:3 dilution in 50 mM HEPES, pH 7.5, 50 mM NaCl, and 50 mM EDTA and incubation at room temperature for 5 min. Chase reactions were then initiated by mixing 200 nM UBE2L3~*Ub(K0) with 200 nM TRIP12 (or TRIP12 variants) and acceptor. Unless noted otherwise, WT full-length TRIP12 was used for assays. Specific acceptors and their concentrations for each experiment are reported below, in order of appearance in the main text. Nomenclature for mutant K48-linked di-Ubs is delineated in the section describing their generation. Where no mutations are indicated, native K48 di-Ub from in vitro K48 chain formation reactions was used (see previous section).
Determination of substrate di-Ub linkage specificity (Fig. 1a) was performed using 200 nM or 1 µM of mono-Ub or di-Ub of designated linkage types (purified from in vitro chain formation reactions or synthesized as specified in previous section) as the acceptor.
For the substrate concentration dependence assays (Extended Data Fig. 1a), K48 di-Ub and mono-Ub were added at 0.2, 0.5, 1, or 2 µM, as indicated. Negative control reactions contained the corresponding K29R variants at 2 µM.
The preference for proximal over distal Ub (Fig. 1b) was characterized by testing the activity of TRIP12 on 200 nM of UbK48R–UbD77, UbK48R–UbK29R D77, UbK29R K48R–UbD77 or UbK29R K48R–UbK29R D77.
The influence of acceptor side chain length was tested by comparing TRIP12 activity on 200 nM of semi-synthetic UbK29R K48R–UbK29Dap D77 (C1), UbK29R K48R–UbK29Dab D77 (C2), UbK29R K48R–UbK29Orn D77 (C3), UbK29R K48R–UbD77 (C4), UbK29R K48R–UbK29hLys D77 (C5), or fully recombinantly produced UbK29R K48R–UbD77 (C4 bio). Band intensities of tri-Ub products at 7-s timepoints were quantified using ImageJ72 and plotted using GraphPad Prism 10.
The truncated TRIP12ΔN variant was validated (Extended Data Fig. 1c) by comparing the activity of 200 nM TRIP12 or TRIP12ΔN on 200 nM K48 di-Ub or mono-Ub, and the corresponding UbK29R variants were used as negative controls.
Assays showing the loss of activity in TRIP12 point mutants (Figs. 3b and 4a–c and Extended Data Fig. 8a) were performed using 200 nM full-length TRIP12 (carrying the indicated substitutions or WT as positive control), and 200 nM K48 di-Ub or 1 µM mono-Ub as the acceptor.
To test the effects of substitutions in a K48 di-Ub acceptor (Figs. 3b and 4b), 200 nM of UbK29R K48R–UbD77 (‘WT’ in legend), UbK29R K48R–UbR42A D77, UbK29R K48R–UbR42E D77, UbK29R K48R–UbE16K D77, UbK29R K48R–UbD21A D77 or UbK29R K48R–UbN25D D77 was added to chase reactions, together with 200 nM WT TRIP12. For substitutions in the mono-Ub acceptor (Extended Data Fig. 8a), 1 µM Ub–6×His or the corresponding variants were used instead.
In assays testing TRIP12 activity on mono-, K29 tetra-, and K48 tetra-Ub with different equivalents of UBE2L3~*Ub(K0) (Extended Data Fig. 9a), two pulse reactions with 15 µM or 75 µM *Ub(K0) and 12 µM or 60 µM UBE2L3 (for 1 and 4 equivalents, respectively) were set up and further processed as described above, such that final assay concentrations were 1 µM or 4 µM UBE2L3~Ub; 1 µM of mono-Ub or K29 tetra-Ub or 250 nM K48 tetra-Ub was used as the acceptor in chase reactions.
Assays comparing K29R and WT donor in the modification of mono-, K29 tetra-, and K48 tetra-Ub by TRIP12 (Extended Data Fig. 9b) were performed using fourfold excess of UBE2L3~Ub. Pulse reactions were performed as described above, but using the indicated untagged Ub variants (K29R or wild type) as donors. Acceptors were added to chase reactions at a final concentration of 1 µM for mono- and K29 tetra-Ub or 250 nM for K48 tetra-Ub.
For the DUB cleavage assay to distinguish products on the basis of susceptibility to OTUB1* (a K48-linkage-specific DUB), both alone and in combination with TRABID (a K29-linkage-specific DUB) (Extended Data Fig. 9c), reactions were set up with the indicated acceptor, consisting of mono-Ub (1 µM), K29-linked tetra-Ub (1 µM), and K48-linked chains (250 nM), including 4 equivalents of UBE2L3~Ub (untagged wild type), as described in previous section. The mixtures were incubated for 5 min, then quenched by addition of 500 mM HEPES, pH 7.5, 1.5 M NaCl, and 50 mM DTT (10 µl in 80 µl reaction). Reactions were split into three 20-µl portions, and 2.5 µl of OTUB1* (1 µM final concentration) or DUB dilution buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, and 10 mM DTT) were added. Reactions were incubated at 37 °C for 30 min, followed by the addition of 2.5 µl TRABID (500 nM final concentration) or DUB dilution buffer and further incubation at 37 °C, then were quenched by the addition of reducing SDS–PAGE sample buffer.
In assays comparing mutants that distinguish chain formation by different linkage-specific HECT E3s (Fig. 5d, left), chase reactions contained 200 nM TRIP12 and 1 µM Ub–6×His, UbA46F–6×His, or UbD21A–6×His. Corresponding experiments with UBR5 (Fig. 5d, right) were performed using 200 nM UBE2D2~*Ub (as E2~Ub, generated as described in ref. 9), 200 nM UBR5, and 2 µM Ub-6×His or variants.
Samples were collected at the indicated timepoints and quenched by mixing with SDS–PAGE sample buffer (final concentration: 50 mM Tris, pH 6.8, 10% glycerol, 15 mM EDTA, 0.5% bromophenol blue, 2 % SDS; for reducing, 100 mM DTT was added), then analyzed by SDS–PAGE on 6–22% gels. Gels of assays using fluorescent *Ub(K0) as the donor were scanned using an Amersham Typhoon 5 (Cy2 channel), followed by staining with Coomassie Brilliant Blue to visualize protein inputs. Gels of assays containing untagged WT and K29R donor Ubs were subjected to immunoblotting using anti-Ub P4D1 antibody (Cell Signaling Technology, cat. no. 14049S, diluted 1:5,000 in 5% milk in TBS-T), developed using Pierce ECL substrate (Thermo Scientific, cat. no. 32209), and imaged on an Amersham Imager 600 (GE Lifesciences). Blots were denatured after transfer by incubation in 6 M guanidinium hydrochloride for 30 min. All displayed gels and blots are representative of at least two independent technical replicates.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Comments (0)