
Remember me


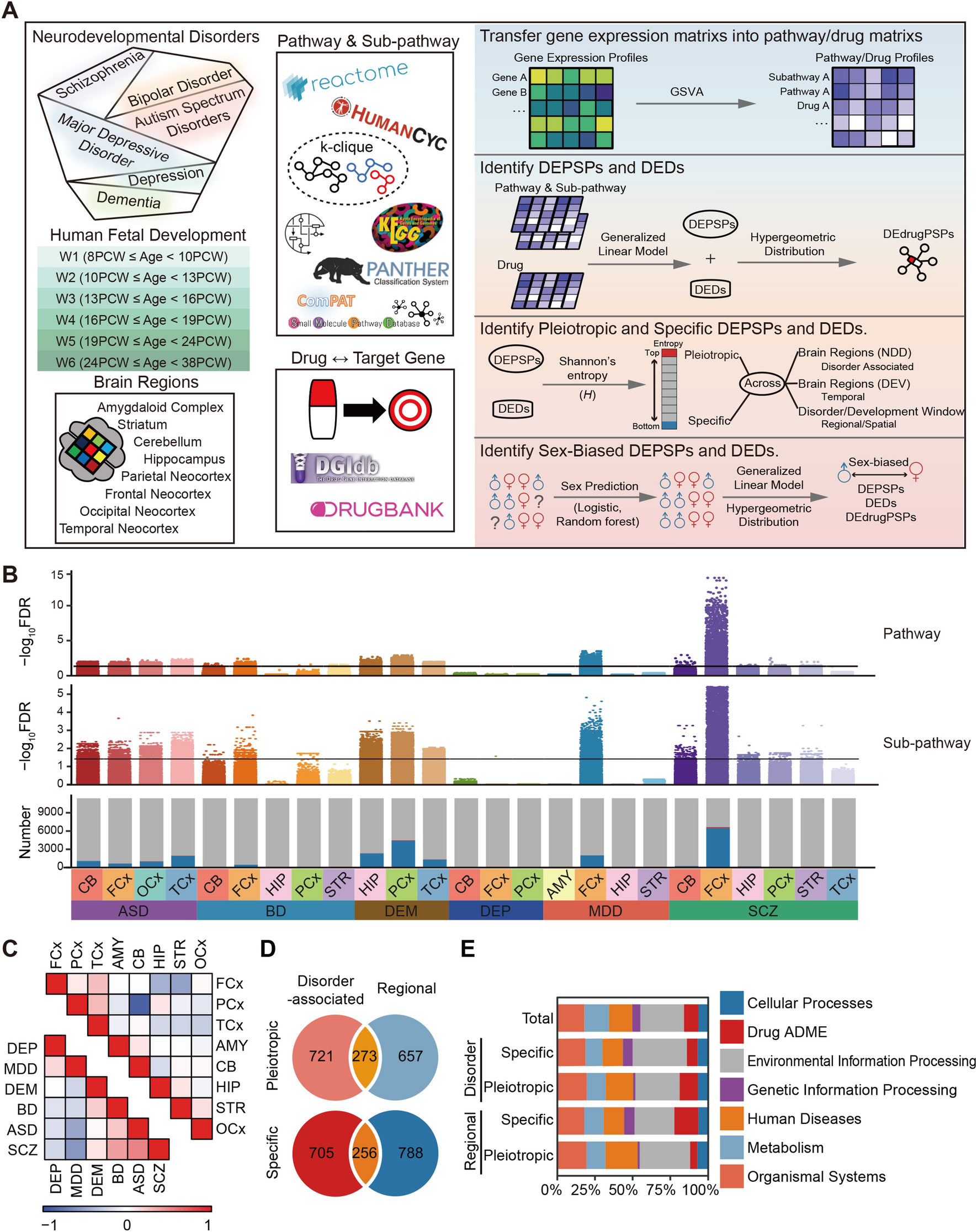

The proband is a 7-year-old boy (Fig. 1A, B). At 4 years old, he exhibited developmental delay, particularly in fine motor skills and expressive language. In addition, he displayed impaired social interaction, stereotypic repetitive behaviors, and hyperactivity, and was diagnosed with autistic spectrum disorder (ASD) and attention deficit hyperactivity disorder (ADHD). Brain magnetic resonance imaging (MRI) revealed a severe deficit in myelin, with a low-normal myelin signal in the left ventricular posterior horn in T2-weighted images (Fig. 1C). By the age of 5, there was an improvement in his linguistic competence. However, in school, he exhibited social difficulties, accompanied by behavioral issues such as sudden yelling and disobeying instructions. At 7 years old, ASD and ADHD persisted.
Fig. 1
TMEM63A_A632T is a loss-of-function mutation. A Sanger sequencing around the mutant site in the proband and his parents. B Family pedigree of the rare variant. Black symbols represent individuals carrying the rare variant, while white symbols represent individuals without the variant. The circle denotes women, and the squares denote men. C MRIs (axial T2-weighted images) of the patient’s brain, which showed abnormal signals in the left ventricular posterior horn (orange arrows). D Sequence alignment of TMEM63A around A632 among species. E Structure of TMEM63A protein based on Zheng, et al. [11] (Protein Data Bank: 8ehw) and the location of A632 (spheres). The extracellular (Ex) and intracellular (In) surfaces of the plasma membrane are indicated by gray lines. The pore-lining transmembrane helices are shown in blue. F Fluorescent emission images of GCaMP6f-positive cells in response to 170 mOsm/L hypotonic solution. White arrowheads indicate the responding cells. Scale bar, 100 µm. G Ca2+ fluorescence intensity traces of cells expressing TMEM63A-P2A-GCaMP6f responding to 170 mOsm/L solution in a representative experiment. H The maximal responses of cells pooled from three experiments challenged by 170 mOsm/L solution plotted versus the exposure time. Red, Ca2+ fluorescence increases >50% are considered to be positively-responding cells. I Ca2+ fluorescence intensity traces of cells expressing TMEM63A_A632T-P2A-GCaMP6f in response to 170 mOsm/L solution in a representative experiment. J The maximal responses of cells pooled from three experiments challenged by 170 mOsm/L solution versus the exposure time. K Averaged Ca2+ fluorescence changes upon hypotonic challenge (Hypo). L Statistical analysis of the maximal Ca2+ rise in response to 170 mOsm/L solution. Cells transfected with TMEM63A_A632T have a lower Ca2+ transient (***P <0.001, t-test; hTMEM63A: n = 186 cells; A632T: n = 337 cells). M Percentage of cells responding to 170 mOsm/L solution. The responding ratio is significantly reduced in A632T compared with WT TMEM63A (****P <0.0001, t-test; n = 3 experiments). The number of cells tested and responding (in brackets) is indicated above the bars.
To explore possible causal genetic variants, we conducted exome sequencing (ES) on the individual, revealing a heterozygous missense variant of TMEM63A (GenBank: NM_014698.3; c.1894G>A, p.[Ala632Thr]). In the general population of ExAC and gnomAD databases, the allele frequency of the c.1894G>A variant is 0.0008249% and 0.001591% respectively. Notably, this variant was conspicuously absent in controls of East Asian descent within both databases. The affected amino-acid position, p.Ala632, is conserved (positive scores in PhyloP: 4.077). The missense variant p.Ala632Thr was predicted to be damaging by the functional prediction algorithms combined annotation-dependent depletion (CADD) (Phred Score = 28.7), deleterious annotation of genetic variants using neural networks (DANN) (Score: 28.7), and likelihood ratio test (LRT) (Score: 0.00). No other clinically relevant deleterious mutations were found.
Sanger sequencing was applied to analyze the locus for the parents, revealing that the rare variant in the proband was inherited from his father (Fig. 1A, B). Notably, the father displayed a sluggish response in verbal communication and disclosed a history of delayed language development and limited verbal interactions during his youth. This background information suggested the possibility of developmental delay, particularly in language, for the father. This comprehensive analysis led us to hypothesize that the heterozygous mutation in TMEM63A is most likely responsible for the myelin sheath abnormality in the patient. This variant impacts an amino-acid situated within the transmembrane domain (M8) (Fig. 1D). The residue is alanine in the human and chimpanzee, but valine in other animals including the mouse, chicken, frog, and zebrafish. Interestingly, valine is also present in human TMEM63B and TMEM63C. Despite the evolutionary transition from valine to alanine in some mammals, both valine and alanine share common characteristics as hydrophobic residues with small side chains. The patient’s mutation introduces a hydrophilic residue, threonine, in place of the original alanine, potentially disrupting the channel function (Fig. 1E) [11].
TMEM63A_A632T Is a Loss-of-function MutationTo study the effect of this mutation on TMEM63A function, we cloned human TMEM63A and constructed TMEM63A-P2A-GCaMP6f in the pCAGGs vector. The construct was transfected into N2a cells, allowing separate expression of TMEM63A and the Ca2+-sensitive reporter GCaMP6f. A hypotonic solution (170 mOsm/kg) was applied to activate TMEM63A and Ca2+ fluorescence was monitored (Fig. 1F) [14]. The [Ca2+]i elevation was significantly lower in N2a cells transfected with TMEM63A_A632T than in those with wild-type TMEM63A (Fig. 1G–L). In addition, the ratio of responding cells among the total population was markedly reduced (Fig. 1M). These findings strongly suggest that the A632T variant represents a LoF mutation in TMEM63A.
TMEM63A Is Abundantly Expressed in OLs in the BrainWe then sought to study the function of TMEM63A in myelination. To visualize the expression of TMEM63A in the brain, we generated a Tmem63aEGFP knock-in reporter mouse line. In this model, a loxP-EGFP-polyA-loxP sequence was inserted in front of the start codon of the endogenous Tmem63a gene (Fig. 2A). Consequently, EGFP expression was driven by the Tmem63a promoter, providing insight into the TMEM63A expression pattern. Double staining for Olig2 and GFP (Fig. 2B) revealed that the majority of GFP+ cells in the cortex (~91.2%) were Olig2-positive at P14 (Fig. 2C), indicating predominant expression of TMEM63A in OL lineage cells. Within this population, 3.7% were co-stained with Pdgfrα, a marker of OPCs, while the remaining cells co-stained with CC1, indicating differentiated OLs (Fig. 2D).
Fig. 2
TMEM63A is abundantly expressed in oligodendrocytes in the brain. A Generation of Tmem63aEGFP mice. B Co-immunostaining of GFP with Olig2, Pdgfrα, or CC1 in the cortex of Tmem63aEGFP at P14. The panels on the right are enlarged from the boxed areas. The white arrowheads indicate the double positive cells. Scale bars, 50 μm. C Numbers of Olig2+ cells among GFP+ cells in the cortex at P14. D Numbers of CC1+ or Pdgfrα+ cells among GFP+ cells in the cortex at P14.
We further examined EGFP expression in the brains of Tmem63aEGFP mice at different developmental stages. Western blots of whole brain lysis showed developmental increases in the EGFP expression in the Tmem63aEGFP mice. Interestingly, the EGFP expression is nicely coordinated with the expression of Mbp, indicating that TMEM63A expression is increased with OL differentiation and maturation (Fig. S1A, B).
Loss of TMEM63A Disrupts Proper MyelinationTo investigate the function of TMEM63A in the brain, we used a CRISPR-Cas9-based gene targeting strategy to delete Tmem63a in mice (Fig. 3A). No detectable Tmem63a mRNA in the brains of 2-month Tmem63a−/− mice indicated successful deletion of Tmem63a (Fig. 3B). At the time of birth, Tmem63a−/− mice were visually indistinguishable from wild-type littermates (WTs), showing no defects in suckling. However, Tmem63a−/− mice were notably smaller than WTs at P14 (Fig. 3C). These pups displayed a significant reduction in weight (Fig. 3D) and length (Fig. 3E). Furthermore, the brain size was notably smaller (Fig. 3F, G).
Fig. 3
Loss of TMEM63A disrupts proper myelination. A Generation of Tmem63a knockout mice: Exon2 of TMEM63A is completely deleted. B qRT-PCR analyses of Tmem63a in the brains of 2-month-old Tmem63a−/− and WT mice (****P <0.0001, t-test; n = 6 mice per genotype). C–E Body size of WT and Tmem63a−/− mice (C). The body weight (D; P = 0.0043, t-test) and body length (E; P = 0.0082, t-test) are lower in Tmem63a−/− mice than in WTs at P14 (WT, n = 4 mice; Tmem63a−/−, n = 3 mice). F Whole brains of WT and Tmem63a−/− mice at P14. G Analysis shows a severe reduction in the size of the brain in Tmem63a−/− mice (P = 0.0216, t-test; n = 3 mice per group). H Western blots for Mag and Mbp. Lysates are prepared from the cortex in WT and Tmem63a−/− mice at P14. β-Tubulin is used as the loading control. I, J Fold change in Mag (P = 0.0028, t-test) and Mbp (P = 0.0028, t-test) between WT and Tmem63a−/− mice (n = 6 mice per group). K Representative images for fluorescence IHC of Mag and Mbp. Scale bar, 100 μm. L, M Ratio of the immuno-reactivity area to total area. The immuno-reactivity of Mag (P = 0.0074, t-test) or Mbp (P = 0.0166, t-test) in the cortex in the Tmem63a−/− mice at P14 (n = 4 mice per group). N TrueGold myelin staining in sections from WT and Tmem63a−/− mice at P14. The myelin sheath tracts in the cortex are sparse in Tmem63a−/− mice. The panels on the right are enlarged from the boxed areas. Scale bars, 100 μm. O Electron microscopic analysis of the myelin sheath. The panels on the right are enlarged from the boxed areas. Scale bars, 2 μm. P The number of myelinated axons per 100 μm2 is significantly lower in Tmem63a−/− mice (P = 0.0058, t-test; n = 3 mice per group). Q Quantification of myelin sheath thickness and the bar graph display the g-ratios of all axons as a function of axonal diameter. The g-ratio is significantly higher in Tmem63a−/− mice (P = 0.0010, t-test; n = 3 mice per group). Data are presented as the mean ± SEM and analyzed with unpaired Student’s t-test. *P <0.05, **P <0.01.
To find out whether the myelin sheath is affected in Tmem63a−/− mice, we made Western blots of myelin-related proteins using cortical (Fig. 3H) and corpus callosum (Fig. S2A) tissues from P14 mice. The levels of Mag and Mbp proteins were significantly reduced in the cortex (Fig. 3I, J) and corpus callosum (Fig. S2B, C) of Tmem63a−/− mice compared with WTs. Consistent with this, immunofluorescence (IF) showed significantly decreased Mag and Mbp signals in the cortex (Fig. 3K–M) and corpus callosum (Fig. S2D–F) of Tmem63a−/− mice. TrueGold myelin staining demonstrated a reduction in positive signals in the brain slices from Tmem63a−/− mice compared to those from WTs in the cortex (Fig. 3N) and corpus callosum (Fig. S2G). Lastly, we performed an ultra-structural analysis using TEM (Fig. 3O) [3]. The number of myelinated axons was reduced in Tmem63a−/− mice compared with WTs at P14 (Fig. 3P). In addition, the ratio of the diameter of an axon to the diameter of the axon and myelin sheath (g-ratio) was significantly larger in Tmem63a−/− mice than in WTs (Fig. 3Q), suggesting that the myelin sheath is thinner in Tmem63a−/− mice. Taken together, the above results suggest that Tmem63a deficiency leads to hypomyelination in the brain.
Loss of TMEM63A Impedes OL Differentiation in the BrainTo determine the cellular mechanisms underlying the hypomyelination phenotypes in Tmem63a−/− mice, we analyzed OL lineage cells using brain sections at P14. We found no difference in the density of Olig2+ cells in the cortex (Fig. 4A, B) and corpus callosum (Fig. S3A, B) between WT and Tmem63a−/− mice, suggesting that loss of TMEM63A does not affect the total population of OL lineage cells in the brain. However, the density of Pdgfrα+/Olig2+ cells in the cortex (Fig. 4A, C) and corpus callosum (Fig. S3A, C) of Tmem63a−/− mice was higher than that in WTs at P14. In contrast, the density of CC1+/Olig2+ cells in Tmem63a−/− mice was significantly lower in the cortex (Fig. 4A, D) and corpus callosum (Fig. S3A, D) than in WTs. To explore whether the neuron or microglia affect myelin development, we performed immunofluorescence staining experiments on NeuN (Fig. 4E) and Iba1 (Fig. 4H). We found that the density of NeuN+ cells (Fig. 4F) and Iba1+ cells (Fig. 4I) were unaltered in the Tmem63−/− mice compared with WT mice. Consistent with this, IF on brain sections at P14 indicated that the immunoreactivity of NF200 in Tmem63a−/− mice was comparable to that in WTs (Fig. 4G). Overall, these results suggest that deletion of TMEM63A does not change the population of neurons or microglia. The reduction of CC1+/Olig2+ cells and the increase of Pdgfrα+/Olig2+ cells may be indicative of the failure of OPCs to differentiate into OLs at P14 in Tmem63a−/− mice. To test this possibility, we performed bromodeoxyuridine (BrdU) birth-dating experiments [3]. BrdU was intraperitoneally injected into mice at P4 for six consecutive days and brain sections were prepared at P14 (Fig. 4K). BrdU+/CC1+ cells were rarely detected in the cortex (Fig. 4J) and corpus callosum (Fig. S3E) of Tmem63a−/− mice. Compared to WTs, the density of BrdU+/CC1+ cells was significantly decreased in the cortex (Fig. 4L) and corpus callosum (Fig. S3F) of Tmem63a−/− mice. These data demonstrated that OL differentiation is impaired in Tmem63a−/− mice at P14.
Fig. 4
Loss of Tmem63A impedes oligodendrocyte differentiation in the brain. A Representative images for IF of Olig2, Pdgfrα/Olig2, and CC1/Olig2. Images are from the cortex of control and Tmem63a−/− mice at P14. B The density of Olig2+ cells in the cortex (P = 0.3279, t-test; n = 5 mice per group; ns, no significance). C The density of Pdgfrα+/Olig2+ cells in the cortex (P = 0.0015, t-test; n = 5 mice per group). D The density of CC1+/Olig2+ cells in the cortex (P = 0.0075, t-test; n = 5 mice per group). E Representative images for IF on NeuN. Images are from the cortex of control and Tmem63a-/- mice at P14. F The density of NeuN+ cells in the cortex (P = 0.3304, t-test; n = 5 mice per group). G Representative images for fluorescence IHC on NF200 and Mbp. Scale bar, 100 μm. H Representative images for IF on Iba1. Images are from the cortex of control and Tmem63a-/- mice at P14. I The density of Iba1+ cells in the cortex (P = 0.8770, t-test; n = 6 mice per group). J Representative images of double-staining for BrdU/CC1 in the cortex. The panels on the right are enlarged from the boxed areas. K Experimental paradigm for BrdU injection in the cortex of mice. BrdU is injected once a day for six days from P4 to P9. Brain sections are cut at P14. L The number of BrdU+/CC1+cells per mm2 in the cortex (P = 0.0006, t-test; n = 6 mice per group). M Representative images of double-staining for Olig2/BrdU. The panels on the right are enlarged from the boxed areas. N Experimental paradigm for BrdU injection in the cortex of mice at P4. BrdU is injected for 2 h before cutting sections. O Numbers of BrdU+/Olig2+ cells per mm2 (P = 0.4286, t-test; n = 3 mice per group). P Representative images of double-staining for Olig2/Ki67 in the cortex. Sections from the control and Tmem63a−/− mice at P14 are used. The panels on the right are enlarged from the boxed areas. Q Ratio of Ki67+/Olig2+ cells to Olig2+ cells (P = 0.8130, t-test; n = 5 mice per group). R Representative images of double-staining for CC3/Olig2 in the cortex. Sections from the control and Tmem63a−/− mice at P14 are used. The panels on the right are enlarged from the boxed areas. S Number of CC3+/Olig2+ cells per mm2 (P = 0.4306, t-test; n = 6 mice per group). Scale bars, 50 μm in A, E, H, J, M, P, and R, and 100 μm in G. ns, not significant; **P <0.01, ***P <0.001.
The increase of Pdgfrα+/Olig2 + cells in Tmem63a−/− mice could also result from enhanced OPC proliferation. To examine this possibility, we injected BrdU into pups at P4 and collected brain sections 2 h later (Fig. 4N). The density of BrdU+/Olig2+ cells in the cortex (Fig. 4M, O) and corpus callosum (Fig. S3G, H) did not differ between WT and Tmem63a−/− mice. Furthermore, co-staining with Ki67/Olig2 showed no significant difference in the ratio of Ki67+/Olig2+ cells to Olig2+ cells in the cortex (Fig. 4P, Q) and corpus callosum (Fig. S3I, J) between WT and Tmem63a−/− mice at P14. These experiments thus demonstrated that deletion of TMEM63A does not affect the proliferation of OPCs in the CNS. The increase of Pdgfrα+/Olig2+ cells in Tmem63a−/− mice is more likely to be due to failure to differentiate into OLs.
The reduction of CC1+/Olig2+ cells may also be caused by the apoptosis of OLs. Thus we studied apoptosis by staining cleaved caspase3 (CC3) using brain sections at P14 (Fig. 4R). There was no difference in the density of CC3+/Olig2+ cells in the cortex between WT and Tmem63a−/− mice (Fig. 4S), and CC3+/Olig2+ was hardly observed in the corpus callosum (Fig. S3K), suggesting that deletion of TMEM63A does not cause abnormal apoptosis in OL lineage cells in the CNS. The decrease of CC1+/Olig2 + cells in Tmem63a−/− mouse is more likely due to reduced differentiation.
Myelin in Tmem63a −/− Mice Is Normal at P28Several studies have shown that missense mutations in the TMEM63A gene result in temporary deficient myelination in the brain of infants [29,30,31]. To explore whether such hypomyelination is also transient in Tmem63a−/− mice, we first conducted a Western blotting analysis on myelin-related proteins (Fig. 5A), using cortical tissues from P21 mice. We found that the expression levels of Mag and Mbp remained decreased in the cortex of Tmem63a−/− mice relative to the WTs (Fig. 5B, C). IF (Fig. 5D) and TrueGold myelin staining (Fig. 5E) confirmed these findings, with fewer myelin sheath fibers in P21 Tmem63a−/− mice than in WTs. However, there was no difference in the density of Olig2+ cells (Fig. 5F, G), Pdgfrα+/Olig2+ cells (Fig. 5F, H), or CC1+/Olig2+ cells (Fig. 5F, I) in the cortex of Tmem63a−/− mice compared to WTs. The above results suggested that myelin abnormalities, though partially recovered, persist in the cortex of Tmem63a−/− mice at P21.
Fig. 5
Myelin abnormalities exist in Tmem63a−/− mice at P21. A Western blots for Mag and Mbp. Lysates are prepared from the cortex in WT and Tmem63a−/− mice at P21. β-Tubulin is used as the loading control. B, C Fold change of protein levels for Mag (B) (P = 0.0359, t-test) or Mbp (C) (P = 0.0254, t-test). n =5 mice per group. D Representative images for fluorescence IHC on Mag and Mbp. Scale bar, 100 μm. The immuno-reactivity on Mag or Mbp is low in the cortex in the Tmem63a−/− mice at P21. E TrueGold myelin staining. Brain sections from WT and Tmem63a−/− mice at P21 are used. The panels on the right are enlarged from the boxed areas. Scale bars, 100 μm. F Representative images for IF on Olig2, Pdgfrα/Olig2, and CC1/Olig2. Images are from the cortex of control and Tmem63a-/- mice at P21. Scale bar, 50 μm. G The density of Olig2+ cells in the cortex (P = 0.4176, t-test; n = 5 mice per group). H The density of Pdgfrα+/Olig2+ cells in the cortex (P = 0.2569, t-test; n = 5 mice per group). I The density of CC1+/Olig2+ cells in the cortex (P = 0.3247, t-test; n = 5 mice per group). *P <0.05; ns, not significant.
Western blot analysis on corpus callosum samples from P21 mice (Fig. S4A) showed no significant differences in the levels of Mag and Mbp between control and Tmem63a−/− mice (Fig. S4B, C). IF of Mag and Mbp on brain sections at P21 in the corpus callosum of Tmem63a−/− mice were comparable to that of the controls (Fig. S4D). TrueGold myelin staining showed there was no significant difference in the density of TrueGold-positive signals in the corpus callosum of Tmem63a−/− mice compared with WTs (Fig. S4E). Finally, there was no difference in the density of Olig2+ cells (Fig. S4F, G), Pdgfrα+/Olig2+ cells (Fig. S4F, H), and CC1+/Olig2+ cells (Fig. S4F, I) in the corpus callosum. These data suggested that the myelin is recovered in the corpus callosum by P21.
By P28, there were no significant differences in body weight (Fig. 6A, B) and body length (Fig. 6A, C) between Tmem63a−/− mice and WTs. Meanwhile, the brain weights of Tmem63a−/− and WT mice were compared (Fig. 6D, E). We subsequently assessed the expression of myelin proteins in Tmem63a−/− mice in comparison to controls at P28. Western blot analysis was applied to cortical samples from P28 mice (Fig. 6F), revealing no significant differences in the levels of Mag and Mbp (Fig. 6G, H). We then applied IF to brain sections at P28 (Fig. 6I), which indicated that the immunoreactivity of Mag and Mbp in Tmem63a−/− mice was comparable to that of the controls (Fig. 6J, K). TrueGold myelin staining showed there was no significant difference in the density of TrueGold-positive signals between Tmem63a−/− mice and WTs (Fig. 6L). Further, TEM demonstrated that the density of myelinated axons (Fig. 6M, N) and the thickness of the myelin sheath (Fig. 6M, O) were similar in control and Tmem63a−/− mice at P28. Finally, we applied IF to OL lineage cells using several markers at P28 (Fig. 6P). There was no difference in the density of Olig2+ cells (Fig. 6Q), Pdgfrα+/Olig2 + cells (Fig. 6R), or CC1+/Olig2 + cells (Fig. 6S) in the cortex between WTs and Tmem63a−/− mice. Collectively, these results indicate that, although the expression of myelin proteins was significantly reduced at P14, it had fully recovered by P28. These findings suggest that the TMEM63A protein may be a critical regulator that determines the timing of OL differentiation during development.
Fig. 6
Myelin in Tmem63a−/− mice is normal at P28. A–C Body size of WT and Tmem63a−/− mice. The body weight (B) (P = 0.0689, t-test) and the length of the body (C) (P = 0.0825, t-test) are not significantly different between Tmem63a−/− mice and WTs at P28 (n = 6 mice per group). D Whole brains of WT and Tmem63a−/− mice at P28. E Quantitative analysis showing no significant difference in the size of the brain between Tmem63a−/− mice and WTs at P28 (P = 0.2058, t-test; n = 3 mice per group). F Western blotting for Mag and Mbp. Lysates are prepared from the cortex in WT and Tmem63a−/− mice at P28. β-Tubulin is used as the loading control. G, H Fold change of protein levels. There are no significant differences in Mag (G) (P = 0.5235, t-test) and Mbp (H) (P = 0.7869, t-test) between WT and Tmem63a−/− mice (n = 5 mice per group). I Representative images of IF for Mag and Mbp. Scale bar, 100 μm. J, K Ratio of the immuno-reactivity area to total area. The immuno-reactivity for Mag (J) (P = 0.8880, t-test) or Mbp (K) (P = 0.9829, t-test) does not significantly differ between control and Tmem63a−/− mice at P28 (n = 4 mice per group). L TrueGold myelin staining of sections from WT and Tmem63a−/− mice at P28. The panels on the right are enlarged from the boxed areas. Scale bars, 100 μm. M Electron microscopic images and analysis of the myelin sheath. The panels on the right are enlarged from the boxed areas. Scale bars, 2 μm. N Numbers of myelinated axons per 100 μm2 are comparable between control and Tmem63a−/− mice (P = 0.6305, t-test; n = 3 mice per group). O Myelin sheath thickness and bar graph of g-ratios of all axons as a function of axonal diameter. The g-ratio is comparable in control and Tmem63a−/− mice (P = 0.4370, t-test; n = 3 mice per group). P Representative images of IF for Olig2, Pdgfrα/Olig2, and CC1/Olig2 in the cortex at P28. Scale bar, 50 μm. Q The density of Olig2+ cells in the cortex (P = 0.7151, t-test; n = 5 mice per group). R The density of Pdgfrα+/Olig2+ cells in the cortex (P = 0.6285, t-test, n = 5 mice per group). S The density of CC1+/Olig2+ cells in the cortex (P = 0.3369, t-test; n = 5 mice per group). ns, not significant.
Myelin Dysplasia in Primary Cultured Tmem63a −/− OPCsIn order to further validate the above findings, OPCs were prepared from the cortex of P7 pups and cultured in an oligosphere medium. After 3 days, a mitogen-free medium was applied to promote the differentiation of OPCs. Generally, after culture in the mitogen-free medium for 48 h, processes were extended from the cell body, suggesting that OPCs were differentiating into OLs. The processes then extended and branched, and the OLs became increasingly complex in arborization in vitro. The morphology of OLs (process formation and branching) and the expression of myelin proteins were analyzed after differentiation for 48, 72, 96, and 120 h in vitro. The fluorescence immunohistochemistry of Mbp was significantly lower in differentiated OLs from Tmem63a−/− mice than those from WTs at 48 or 72 h but did not differ at 96 and 120 h (Fig. 7A, B). Besides, the average length of the processes of OLs from Tmem63a−/− mice was significantly shorter than that of the WT OLs at 48 or 72 h but comparable at 96 and 120 h (Fig. 7C, D). These results suggested that the myelin dysplasia caused by Tmem63a deletion is temporary.
Fig. 7
Myelin dysplasia in primary cultured Tmem63a−/− OPCs. A Immunolabeling for Mbp in primary OPCs differentiated for 48, 72, 96, or 120 h. Scale bar, 50 μm. B Normalized Mbp density is significantly lower in Tmem63a−/− OPCs than controls differentiated for 72 h (72 h: P = 0.0413, t-test; 96 h: P = 0.5864, t-test; n = 3 mice per group). C Representative OLs in primary OPCs differentiated for 48, 72, 96, or 120 h illustrated with the method of Sholl analysis. Scale bar, 50 μm. D Sholl analysis of average process length per cell (48 h: P = 0.0052, t-test; 72 h: P = 0.0006, t-test; 96 h: P = 0.4697, t-test; 120 h: P = 0.5408, t-test; n = 3 mice per group). E Immunolabeling for Mbp in primary OPCs under differentiation conditions containing different concentrations of Ca2+ for 72 h. Scale bar, 50 μm. F Fluorescent emission images of OLs differentiated for 20 h, in response to hypotonic stimulation. Scale bar, 100 μm. G Ca2+ fluorescence intensity traces in WT OLs respond to 170 mOsm/L solution in a representative experiment. H The maximal responses of cells pooled from three experiments challenged by 170 mOsm/L solution versus exposure time. Red, Ca2+ fluorescence increases >50% are considered to be positively-responding cells. I Ca2+ fluorescence intensity traces in Tmem63a−/− OLs responding to 170 mOsm/L solution in a representative experiment. J The maximal responses of cells pooled from three experiments challenged by 170 mOsm/L solution versus exposure time. Red, Ca2+ fluorescence increases >50% are considered to be positively-responding cells. K Ca2+ intensity traces of WT and Tmem63a−/− OLs respond to 170 mOsm/L solution in a representative experiment. L The maximal Ca2+ rise in OLs in response to hypotonic stimulation (****P <0.0001, t-test; +/+: n = 335 cells; −/−: n = 333 cells). M Percentage of cells responding to hypotonic stimulation under indicated extracellular [Ca2+] from three experiments. The number of cells tested and responding (in brackets) is indicated above the bars (****P <0.0001, t-test; n = 3 mice per group). *P <0.05, **P <0.01, ***P <0.001; ns, not significant.
TMEM63A Induces Ca2+ Influx in OPC/OL CulturesTMEM63 proteins function as osmosensitive cation channels that regulate extracellular Ca2+ influx, and previous research has indicated that TMEM63A, in particular, acts as an osmosensitive cation channel activated by hypotonic stress, mediating extracellular Ca2+ influx [14]. This prompted us to hypothesize that TMEM63A-mediated Ca2+ influx plays a pivotal role in OL differentiation. To test this possibility, we manipulated the Ca2+ concentration in the OL differentiation medium. Notably, we found Mbp+ cells at 72 h after differentiation in mitogen-free medium treated with 2.5 mmol/L Ca2+ but not 0.5 mmol/L Ca2+ (Fig. 7E), underscoring the importance of Ca2+ influx in OL differentiation.
To further characterize the ionotropic action of TMEM63A in OLs, we cultured OPCs isolated from WT and Tmem63a−/− mice in mitogen-free medium for 20 h. These cells were then loaded with the Ca2+ indicator dye Fluo-4-AM, and changes in cytoplasmic [Ca2+]i were monitored after switching the extracellular osmolarity from 300 to 170 mOsm/L (Fig. 7F). This hypotonic stimulus induced a ~1.6-fold elevation in Ca2+ fluorescence from the isotonic baseline in WT OLs, while Tmem63a−/− OLs exhibited less elevation (Fig. 7G–L). Furthermore, the ratio of responding cells among Tmem63a−/− OLs was significantly lower than the control (Fig. 7M). These results support the conclusion that TMEM63A is a key regulator of Ca2+ influx in OLs during the early stages of OPC differentiation.
TMEM63A_A632T Fails to Rescue MyelinationWe then explored whether expressing human TMEM63A in Tmem63a−/− OPCs rescues their differentiation. Thus, we constructed lentivirus (LV) carrying human TMEM63A (LV-TMEM63A) and infected cultured Tmem63a−/− OPCs. Significant OL differentiation was seen in OPCs infected with LV-TMEM63A cultured for 72 h in a mitogen-free medium (Fig. 8A), in sharp contrast to uninfected OPCs. Conversely, OPCs infected with LV-TMEM63A_A632T showed very limited differentiation by 72 h in a differentiation medium (Fig. 8A). The processes were longer in cells infected with LV-TMEM63A than those with LV-TMEM63A_A632T (Fig. 8B, C). Furthermore, we microinjected LV-TMEM63A into the cortex of Tmem63a−/− mice at P1 (Fig. 8D) and examined the expression of Mbp in the cortex at P14. Immunostaining showed that the immunoreactivity of Mbp was significantly weaker in the GFP fluorescence region of the cortex in mice with LV-TMEM63A_A632T compared to those infected with LV-TMEM63A (Fig. 8E). The density of GFP+/Olig2+/CC1+ cells was also significantly lower (Fig. 8F, G). These data indicate that re-expressing TMEM63A in Tmem63a−/− OLs can rescue their differentiation. The rescue effects of the LoF mutant A632T are much weaker.
Fig. 8
TMEM63A_A632T fails to rescue myelination. A Double-staining of OLs for Mbp and GFP. Scale bar, 50 μm. B Representative images of β-tubulin immunofluorescence in oligodendrocyte cultures at 3 days in vitro (DIV3). Scale bar, 50 μm. C Average sheath length per cell. OLs with TMEM63A_A632T show a shorter branch length than WTs (P = 0.0008, t-test; WT, n = 14; A632T, n = 8). D Experimental paradigm for lentivirus injection in the cortex of Tmem6a−/− mice. E Representative images of Mbp and GFP immunofluorescence. The immuno-reactivity for Mbp is extremely low in the cortex in the Tmem63a−/− mice re-expressing TMEM63A_A632T at P14. The white arrowheads indicate the double positive signaling. Scale bar, 50 μm. F Representative images for immunofluorescence on GFP, Olig2, and CC1. The white arrowheads indicate the GFP+/CC1+/Olig2+ cells. Scale bar, 50 μm. G The ratio of CC1+/GFP+ cells to Olig2+/GFP+ is extremely low in the cortex of the Tmem63a−/− mice re-expressing TMEM63A_A632T at P14 (P = 0.0272, t-test; n = 3 mice per group). *P <0.05, ***P <0.001.
Tmem63a +/− Mice Exhibit Normal MyelinationTo date, hypomyelination-related TMEM63A mutant patients including ours are all heterozygous de novo mutations. Thus, it is of interest to know whether Tmem63a+/− mice could have hypomyelination phenocopying these patients. We thus examined myelin-related phenotypes in the Tmem63a+/− mice. The protein levels of Mag and Mbp were comparable in the cortex between control and Tmem63a+/− mice at P14 (Fig. S5A–C). Furthermore, Mag+ and Mbp+ myelin were unaltered in the Tmem63a+/− mice (Fig. S5D-F). TrueGold myelin staining showed no significant difference between WT and Tmem63a+/− mice (Fig. S5G). Overall, these observations suggest that the myelination appears normal in the Tmem63a+/− mice. These results indicate that Tmem63a genetic dose insufficiency may be more severe in humans than in mice. Alternatively, the mutant TMEM63A may interfere with the healthy copy of
Comments (0)