
Remember me



The clinical PKPD model was developed based on 1710 serum efgartigimod and 1635 serum tIgG concentrations from a total of 84 healthy volunteers in studies NCT03457649 and NCT03334084. This included all available PKPD data from these studies, except for five PK samples that were considered outliers during initial exploratory analysis. 264 PK samples were BQL, and not included in the analysis. Based on initial PKPD modeling analysis, an impact of ADA on the clinical PKPD profiles was not apparent, and these data were included in the analysis.
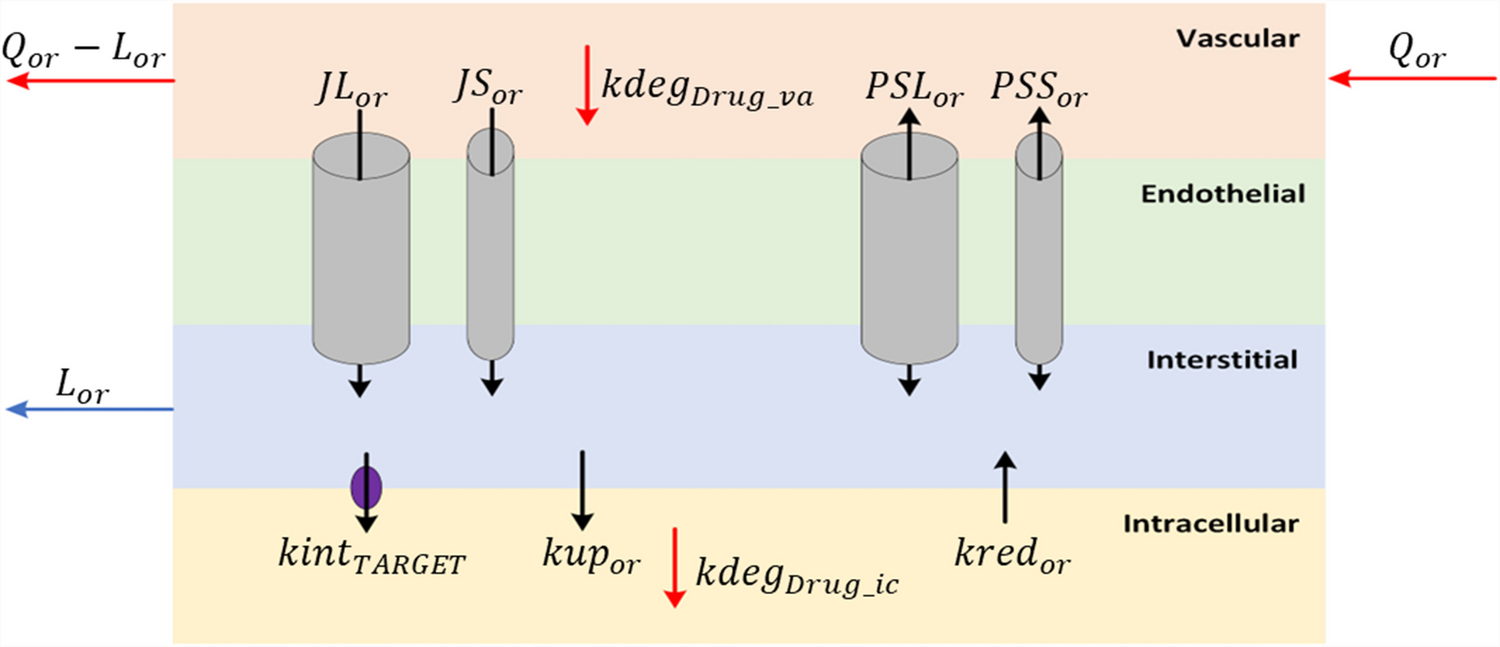
Following an iterative model development process, a clinical PKPD model characterized by FcRn binding was developed with a structure as depicted in Fig. 1 and with parameter estimates and precision presented in Table 1. The NONMEM model code is presented in the Supplementary Material. Efgartigimod entered the central compartment after iv or sc administration. For the latter, a sequential zero order (described by duration D) and first order (described by rate constant ka) process was implemented with an estimated sc bioavailability (F) of 47%. Unbound drug was cleared from the central compartment with a linear clearance CL. Unbound drug distributed to a first and second peripheral compartment with intercompartmental flows Q and Q2 and volumes VP and VP2, respectively. Target binding occurred in the central and first peripheral compartments, with estimated FcRn concentration R0. Target binding was not implemented in the second peripheral compartment. This structure was the result of a data driven approach whereby the fit was worse for alternative models including a two compartmental model with or without peripheral target binding, and a three compartmental model with target binding in both peripheral compartments with different estimated FcRn concentrations. Binding was assumed at steady state with dissociation constant KD, with an estimated in vivo scaling factor: the in vivo KD was 2.1 fold larger than the fixed in vitro KD. The FcRn and complex degradation rate constants, assumed to be equal via a single shared estimate, tended to zero and could not be estimated. Consequently, these parameters were fixed to 0. Therefore, FcRn receptor turnover and efgartigimod elimination via the complex, were not included in the model. The underlying assumption following from this implementation was that interaction with efgartigimod did not alter the dynamics or the capacity of the target. Total IgG was described using a turnover model with estimated synthesis rate kin and degradation rate constant in absence of drug kout. The predicted total RO (central and peripheral compartment combined) at pH 7.4 drove the increase in the tIgG degradation, with estimated parameters Emax and γ according to Eq. 1. This structure was the result of a data driven approach whereby the fit was worse for alternative models including models where only the RO for either the central or the peripheral compartment was driving the effect. IIV was included on ka, D, F, CL (and covariance with VP/VP2), VP/VP2, VC, R0, kin and Emax. A single variance was estimated for the IIV on VP and VP2, since the data supported IIV on those parameters but the model was over-parameterized when separate variances were included.
Table 1 Parameter table for the efgartigimod translation PKPD FcRn binding modelFig. 1
Schematic overview of the efgartigimod translation PKPD FcRn binding model
The fixed allometric scaling factors (Eq. 2; Table 1) were included in the model as a preparation step for subsequent translational PKPD predictions. These relationships were assumed to hold and were not evaluated based on the clinical PKPD data. The FcRn concentration was scaled with a scaling coefficient of 0.25, to mimic anticipated FcRn degradation scaling with coefficient − 0.25 with a body weight independent synthesis rate. During translational model development, this implementation was evaluated.
PK (Figure S1) and PD (Fig. 2) goodness-of-fit plots show that the model was able to adequately describe the clinical PKPD data simultaneously for efgartigimod. All parameters were estimated with good precision (Table 1). The strongest parameter correlation was − 0.95 between VP2 and Q, and the eta shrinkage was larger than 30% only for the IIV on the FcRn concentration (i.e. 37%). Total IgG concentrations were modelled and were, during post-postprocessing, also calculated as a percentage relative to pre-dose for interpretation purposes. From the individual fit plots for PK (Fig. 3; left panel), relative tIgG (Fig. 3; right panel) and tIgG (Figure S2; left panel), it was clear that the model was able to capture the observed PK and PD profiles for all dose groups. NPC evaluation showed that the model was predictive for both the PK and PD since the observed percentiles of the NPCs lied within the 95% confidence intervals of the predicted percentiles (Figure S3). At the later time-points in the individual PK fit plots (Fig. 3; left panel) and for the lower concentrations in the residuals versus prediction plots (Figure S1; bottom right panel), a seeming model under-prediction was noted. This was not considered a bias, but rather the result of data censoring below the limit of quantification. To visualize this process, serum efgartigimod predictions and corresponding NPDEs were obtained for all (and BQL and non BQL) PK samples using the M3 method [35], based on a MAXEVAL = 0 (no parameter re-estimation, with empirical bayes estimate adjustment allowed) model run. Based on this analysis, it is clear that the seeming PK bias was strictly due to data censoring, since this was no longer observed when including the BQL samples (Fig. 4). Parameter re-estimation including the BQL samples led to model instability and was not pursued further.
Fig. 2
Goodness-of-fit of the total IgG serum concentration for the clinical PKPD model. Red line: Loess smooth through data; Dashed line: line of identity (A and B) or line indicating 0 (C and D)
Fig. 3
Individual PKPD fit plots panelled by treatment group for the clinical PKPD model. DV: observations; PRED: population predictions; IPRED: individual predictions. For interpretation purposes, relative change from pre-dose is shown up to a maximum of 150%, excluding three outlying values from the plot. All data points are included in the individual tIgG fit plots (Figure S2; left panel)
Fig. 4
NPDE versus population prediction for the efgartigimod serum concentration for the clinical PKPD model including the prediction for BQL PK samples. Normalized prediction distribution errors (NPDEs) follow a standard normal distribution: it is anticipated that 2.5% of the data should fall above and below the dashed lines at +/- 1.96. The percentages listed in the plot indicate the actual percentages of NPDEs outside these ranges
Translational FcRn binding model for efgartigimodThe translational PKPD model was developed based on 1597 / 194 / 889 / 35 PK samples and 809 / 151 / 492 / 59 PD samples in monkeys / rabbits / rats / mice, respectively. These samples were obtained from 116 cynomolgus monkeys, 23 rabbits, 192 rats and 12 mice. 477 non-clinical PK samples were BQL, and not included in the analysis. PKPD samples were ignored from the time of ADA detection onward within an animal, as an influence of ADA on the PKPD of efgartigimod was apparent for non-clinical data during initial exploratory analysis, and quantification of this influence was outside the scope of the present analysis.
As the overall variability in the non-clinical data was larger than in the clinical data, this was accounted for in the models through the implementation of an estimated factor on the residual error for both the PK and total IgG. In the final translational model, the non-clinical residual variance for PK and tIgG was 6 and 3-fold higher, respectively, than the clinical residual variance.
For monkeys, data after sc administration were available. These data were well described when implementing a first order (without zero order rate) absorption which was 78% faster than for healthy volunteers. The sc bioavailability was estimated to be 81% for monkeys.
In the clinical model, the following parameters were implemented to be body weight dependent: CL, Q, Q2, VC, VP, VP2, R0, kout and Emax. Therefore, this model could be applied directly to predict the PKPD data in the non-clinical species, with only estimating the monkey sc parameters, without re-estimating other parameters. During this step, the in vitro KD for FcRn binding at pH 7.4 was implemented to be species-specific, based on in vitro measurements (Table 1), with fixed in vivo scaling factor (value of 2.1) estimated from the clinical model, thus keeping the in vitro affinity ranking between species intact. The results of initial prediction of non-clinical data based on the scaled clinical model were difficult to interpret, given the large differences in observed baseline tIgG values between species: the observed tIgG values were highest in monkeys (156 µM), followed by humans (63 µM), rabbits (46 µM), rats (11 µM) and mice (0.3 µM), excluding the option to account for these differences purely by allometric scaling. Attempts to estimate a species-specific synthesis rate to account for these differences often resulted in numerical instability, especially due to the very low total IgG levels in mice, versus other species. Therefore, it was decided to fix these synthesis rates for the non-clinical species to result in a predicted baseline corresponding with the median observed baseline total IgG per species. Based on the observed median baseline and the median body weight per species (3.2 kg, 4.14 kg, 0.274 kg and an assumed 0.02 kg, for monkeys, rabbits, rats and mice, respectively), the factor difference versus the human synthesis rate, needed to match these median baselines after accounting for allometric scaling of the IgG degradation rate, were derived and fixed in the model (Table 1). If the scaling assumptions would hold perfectly, this would mean that these factors would all be equal to one. These assumptions appeared to hold between humans (factor of one by definition), rabbits (50% relatively higher kinversus humans) and rats (30% relatively lower kinversus humans). However, for monkeys, the IgG synthesis rate was more than five fold larger than in humans, after accounting for allometric scaling, while for mice, this rate was relatively more than 30 fold lower than in humans. Given that IIV is implemented on the IgG synthesis rate, the model allowed the flexibility for each animal to deviate from the fixed population synthesis rate.
After accounting for unexplained baseline tIgG differences, the prediction of the non-clinical PKPD data was re-evaluated: parameter estimates from the clinical PKPD model were kept fixed (except for sc absorption), and the non-clinical data were predicted via allometric scaling of body weight dependent parameters and by accounting for species-specific in vitro target binding affinity. During this step, some bias was still observed and during subsequent translational model development, species-specific effects on the (non-drug effect) model parameters were allowed in the model where needed to resolve the remaining bias, guided by individual random effect plots. As a result, only a few parameters were species-specific which mainly described unexplained PK differences, while the majority of parameters were shared across species, including the parameters which describe the total IgG lowering effect of efgartigimod. The corresponding added effects were:
1.± 30% lower relative (after accounting for allometric scaling) clearance for rabbits versus monkeys and humans.
2.± 2 fold higher relative clearance for rats and mice (shared estimate) versus monkeys and humans.
3.± 3 and ± 7 fold lower VP/VP2 for rats and mice, respectively, versus other species.
4.± 8 fold higher relative FcRn concentration for monkeys versus other species.
Given the limited rodent PK data, especially in mice, it is difficult to judge whether distribution is truly different in these species, or whether the lower volumes serve as a surrogate to capture other unexplained species differences. In addition, estimating the allometric scaling factors did not significantly improve the fit.
In contrast to all other studies, it was apparent that the tIgG levels increased during the course of the 4-week subchronic toxicity study in rats, which led to a strong bias in the PD profiles for this study following initial prediction. Investigation into the mechanistic reasons, including possible developmental/immunological changes for these rats during the study, was considered beyond the scope of the present analysis. One descriptive study effect was allowed in the model to account for this behaviour, which led to the adequate prediction of the PKPD data within this study, using the translational model. Initially, the translational model was developed without this study. In a final step, data from this study were included and the rodent parameters were re-estimated while all non rodent parameters were fixed. In this model, the slope was estimated, which allowed the tIgG degradation to decrease in function of time for rats in the 4-week subchronic toxicity study according to Eq. 3.
$$\begin&\frac\cr&\quad =_-\left(\:_+\:_\:\times\:\:^\:\right)\cr&\qquad\times\:\left(1+\text\text\text\text\text\times\:\text\:\right)\:\times\:\:\left[tIgG\right]\end$$
(3)
which is a modification of Eq. 2, where the tIgG degradation changes as a function of time t with estimated slope parameter.
After accounting for these differences, and without including descriptive species-specific drug effect differences, the PK and tIgG dynamics were well-described by the translational model. No large remaining differences between species were apparent for the PK (Fig. 5; left panel) and PD residuals (Fig. 5; right panel), and for the individual random effects (Figure S4). The goodness-of-fit plots showed an overall adequate fit of the PK (Figure S5) and PD (Figure S6) for the complete dataset. Based on the individual PK (Fig. 6; left panel), relative tIgG (Fig. 6; right panel) and tIgG (Figure S2; right panel) fit plots for monkeys after single dose (shown specifically because rich PKPD profiles were available for these studies), it was clear that the translational model was able to capture the observed PK and PD profiles for these dose groups. The parameters added during the translational step were estimated with good precision (Table 1) and no relevant parameter correlations were noted.
Fig. 5
Boxplots of the serum efgartigimod (left) and tIgG (right) residuals by species for the translational PKPD model. Box plots show the median (black line at center of box) and its approximate 95% confidence interval (grey area), the inter-quartile range (box), the 1.5x inter-quartile range (whiskers) and outliers (stars) defined as values outside the 1.5x inter-quartile range
Fig. 6
Individual PKPD fit plots for sd data in cynomolgus monkeys panelled by treatment group for the translational PKPD model. DV: observations; PRED: population predictions; IPRED: individual predictions
Fig. 7
Typical healthy volunteer simulation of the bound and unbound efgartigimod concentration after 4 weekly iv doses of 0.1 and 10 mg/kg. Predicted unbound (solid line) and target-bound (dashed line) serum efgartigimod concentration
Comments (0)