
Remember me




The following populations will be defined for efficacy and safety analyses:
• Intent-to-treat population (ITT)
The ITT population is defined as all patients randomised in the trial, regardless of whether they actually received treatment. The treatment group will be analysed as randomised.
• Responding patient population
Responding patient population is defined as all patients randomised in the trial, received allocated protocol treatment and achieved partial/complete remission. The treatment group will be analysed as randomised.
• Safety population
The safety population comprises all patients randomised and having received at least one dose of trial treatment. The treatment group will be analysed as treated.
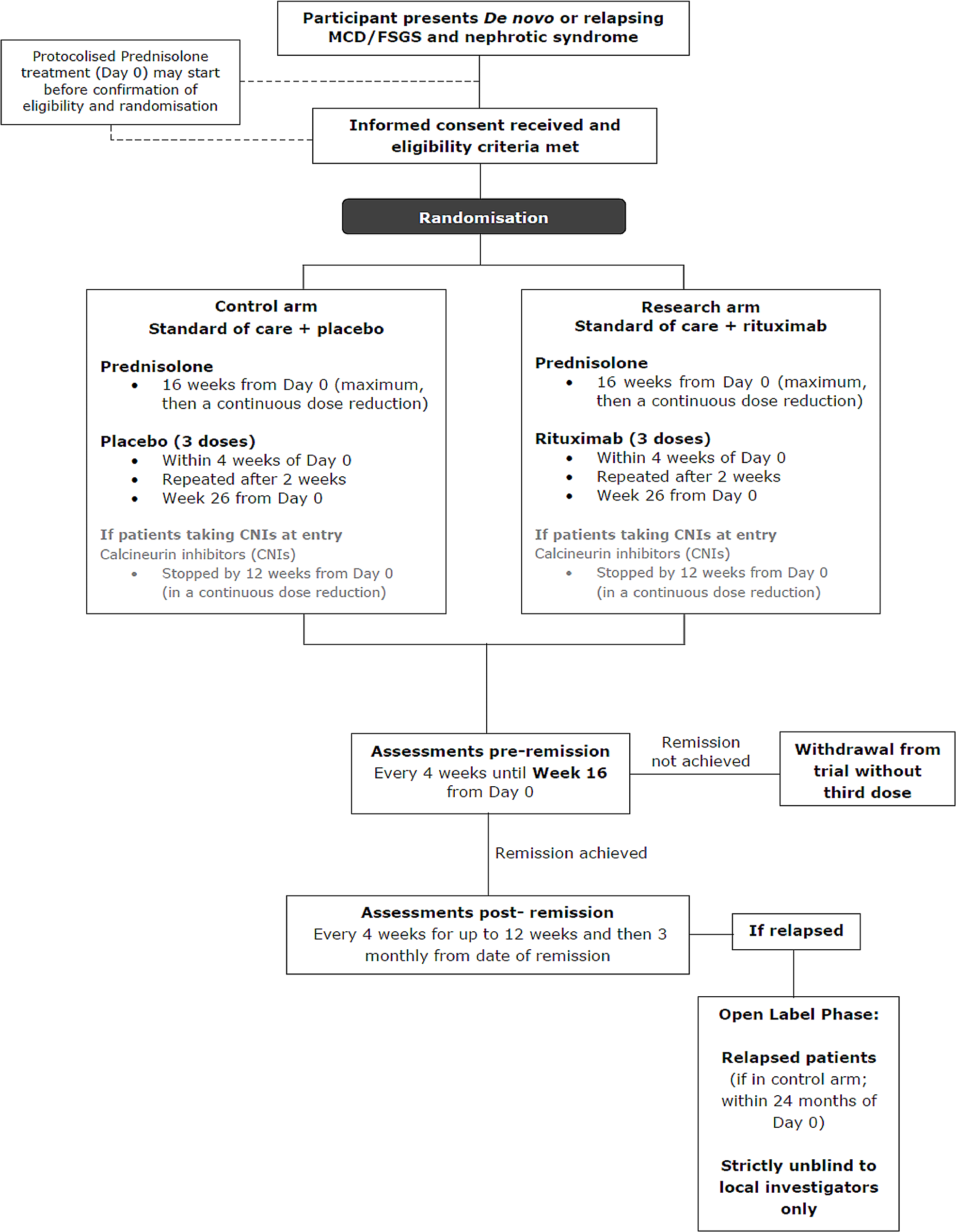
Primary assessment of the efficacy of rituximab at trial endThe primary objective of the trial is to assess the effect of rituximab on the time from remission to relapse of NS. The most clinically relevant benefit is to maintain remission as long as possible whilst minimising steroid toxicity, that is, the duration of remission in responding patients.
If there is no evidence of difference in the distribution of time to remission between the two arms, the stratified log-rank test stratifying the factors used for randomisation will be used.
Otherwise, the probability-of-being-in-response function (PBRF) will be used as a means of estimating the expected duration of remission (EDoR) across all randomised patients given as the estimated response rate times mean duration of remission for responding patients. The analysis will be based on ITT population.
Briefly, considering a stochastic process in which a patient must start in State 0 (that is at Day 0) and eventually progress to an absorbing state 2 (progression, death in the absence of progression, treatment failure at week 16, relapse (or died from disease), possibly passing through a transient state 1 (remission) (Fig. 2). Response and duration of response are assumed to be independent.
Fig. 2
Probability-of-being-in-response function (PBRF)
Summary statistics together with the Kaplan-Meier curves for the duration of remission in responding patients by treatment arm and estimated will also be presented. Detailed statistical analysis methods will be included in the statistical analysis plan.
CensoringFor patients who have achieved partial/complete remission but neither relapsed or died from the disease at the time of the analysis or who are lost to follow up, their data will be censored at the date that they were last known not to have relapsed.
Safety analysisThe safety analyses will be based on the safety population. All safety parameters will be summarised. Summary tables will be presented for incidence rates (number of patients with at least one incidence) of AESI and SAE.
Number of patients to be enrolledThe primary outcome measure is time from remission to relapse from partial/complete remission. With 78 patients randomised at a steady rate over a period of 30 months, who subsequently achieved a partial/complete remission, and an additional 24 months follow-up after the last patient randomised (estimated at 54 months after the first patient was enrolled), the trial will have 80% power (two-sided test, significance level of 5%) to show a 50% change in time to relapse from a median value of 9 months in the control arm to 18 months in the research arm, (i.e. a hazard ratio of 0.50). It is expected that the 67 primary outcome measure events required will have occurred at this point. To achieve 85% power (two-sided test, significance level of 5%), 76 events are required. To achieve 90% power (two-sided test, significance level of 5%), 89 events are required.
With an estimated 75% remission rate, allowing for non-compliance of the order of 5%, a minimum of approximately 112 (112*70%=78.4) patients need to be enrolled, 56 in each arm.
Although the effect size of HR = 0.50 upon which sample size calculations are based appears large, two placebo RCTs of rituximab in children with FR NS have given lower HR at 0.267 (CI 0.135–0.528) [19] and 0.02 (CI 0.01–0.15) [20].
The assumptions used in the sample size estimation are being monitored by the Independent Data Monitoring Committee (IDMC). On the recommendations of the iDMC, the sample size was increased to 130–150, to further inform subgroup analyses by disease subtype (MCD and FSGS).
Economic evaluationCost-effectiveness, cost-utility and value of information analyses will be conducted from the perspectives of NHS and society over the trial duration (within-trial analysis), and over a patient lifetime (decision model-based analyses) comparing rituximab versus placebo.
Cost categories will be drugs (sourced from ‘concomitant medications’ measurement), secondary care contacts (sourced from Hospital Episode Statistics), out of pocket costs and lost productivity accrued by the patient plus carer where applicable (patient questionnaires). The NHS perspective is defined as drugs and secondary care contacts. The societal perspective is defined as NHS plus out of pocket costs and lost productivity.
Outcome for the cost-effectiveness analysis will be adapted from the primary outcome (reported as disease and adverse event free period), and for the cost-utility analysis will be quality-adjusted life years (QALYs), using utilities calculated from the Eq. 5D5L using the recommended valuation set at the time of analysis, and integrated with respect to time.
A decision model will combine evidence from the clinical trial with other relevant evidence obtained from the literature to predict longer term costs and outcomes and hence cost-effectiveness. The model will be used to perform a value of information analysis which will predict the expected return on investment from further research into rituximab in MCD/FSGS.
Ethics and disseminationEthical permission has already been granted:
REC reference: 19/LO/0738, Protocol number: CCTU0228, Version 4.4, 28.09.2023.
IRAS project ID: 258,589. Date of initial first favourable REC Opinion: 14 Jun 2019.
EudraCT number: 2018-004611-50.
ConsentThe investigator or designee ensures that each trial participant is fully informed about the nature and objectives of the trial and possible risks associated with their participation. Written informed consent is obtained from each participant before any trial-specific activity is performed using the ethically approved Informed Consent Form (see supplementary information).
ConfidentialityAll personal information about potential and enrolled participants will be collected, shared, and maintained in order to protect confidentiality before, during, and after the conduct of the trial, according to the requirements of the EU General Data Protection Regulation (GDPR), Data Protection Act 2018 and Cambridge University Hospital NHS Trust policy.
Trial oversight and safetyThe IDMC regularly reviews the data from the trial, including safety and efficacy data. This committee reports to the Trial Steering Committee, who oversee the conduct and progress of the trial.
Adverse event reportingSAEs and other adverse events related to steroids or rituximab are regularly reviewed by Trial Management Group (TMG) and IDMC. Due to the underlying clinical condition of the trial population it is not practicable to report all adverse events in this trial and it is thought that excessive safety reporting may detract from the main objectives of the trial. Rather, only AESI are being reported.
AESIs are as follows:
new onset diabetes mellitus
osteoporosis/fracture
peptic ulceration/gastritis requiring medication/endoscopy
glaucoma
cataracts
weight gain > 10% (not attributed to oedema)
mood changes requiring medication
Patients with NS have frequent hospital admissions related to their underlying disease. Hospital admissions related to hypovolaemia or fluid overload secondary to nephrotic syndrome, or to thromboembolic events, will be exempt from expedited SAE reporting. However, these events will be designated as AESI and details will be collected in the CRFs.
Each Principal Investigator must report all AESIs to the CI using the CRF in a timely manner.
All SAEs should be reported to the CI and TURING trial team using the trial specific SAE form within 24 h of knowledge of the event.
All suspected Adverse Reactions that are related to IMP and that are both unexpected and serious (SUSARs) are subject to expedited reporting. The CI will report all the relevant safety information to the Sponsor, MHRA and the Ethics Committee.
The CI shall inform all investigators concerned of relevant information about SUSARs that could adversely affect the safety of participants.
Dissemination policyOnce completed, the results of TURING will be published in a peer reviewed journal, presented at UK Kidney Week, and data will also be made available on request.
Comments (0)