
Remember me








On the basis of the principles outlined in this Committee Statement, the American College of Obstetricians and Gynecologists (ACOG) offers the following recommendations and conclusions:
All people who could become pregnant, who could have become pregnant, who are lactating, or who identify as women should be presumed eligible for participation in research studies, and their recruitment and participation should be encouraged regardless of race, ethnicity, sexual orientation, gender identity, or socioeconomic status.
Ethical principles mandate fair distribution of benefits and burdens of participation in research, underpinning the need for equitable inclusion in medical research of people who could become pregnant, who could have become pregnant, who are lactating, or who identify as women.
Imposing a heavier burden of consideration on people who could become pregnant, who could have become pregnant, who are lactating, or who identify as women ahead of participation in studies represents a structural barrier to equal inclusion and should be avoided. Respect for autonomous decision making by all people, whether they can become pregnant or not, avoids imposing unethical obstacles to research participation.
A contraception requirement for people who could become pregnant, who could have become pregnant, who are lactating, or who identify as women to participate in pharmaceutical research is paternalistic, discriminatory, and exclusionary. Rather, people may desire counseling about the risks of a potential pregnancy during participation, with the ultimate decision about contraception left to the individual.
In general, requiring participation consent from an intimate partner or nongestational intended parent is not warranted, ethically justified, or legally required in research studies exclusively intended to benefit fetuses.
There is an ethical imperative to increase the inclusion of people who can become pregnant, who are pregnant, and who are lactating in research to improve maternal and fetal health.
A pregnant person in labor is able to undergo the appropriate informed consent process for research, similar to individuals with conditions that may have parallel connotations to labor, including other painful conditions or even those that are life-threatening or emergent (eg, appendicitis, nephrolithiasis, and myocardial infarction).
Appropriate study design can maximize the benefits of enrolling people who can become pregnant, who are pregnant, and who are lactating while minimizing the risks to them, their fetuses, and their neonates.
Generally, pregnant individuals' consent is sufficient for research interventions that affect them or their fetuses, as in other clinical scenarios.
BACKGROUNDHistorically, potential research participants have been excluded from involvement in clinical research because of gender, sex, race, class, immigration status, pregnancy, and sexual orientation. Exclusion because of gender or sex was based on an androcentric model whereby male-typical bodies were understood to be a sufficient stand-in for all adult humans. The National Institutes of Health’s (NIH) 1993 Revitalization Act and changes to the Common Rule attempted to mandate and encourage inclusion in clinical trials of women to address some of these inequities (1); however, little progress has resulted. The unfortunate result is that available data on health, disease, and treatment outcomes for women are severely lacking, hampering the ability to care meaningfully for patients. Concerns about the potential for pregnancy and resulting fetal harm in clinical research participants have led to paternalistic and discriminatory requirements, including 1) mandating contraception and 2) mandating consent from the nongestational intended parent. This document provides a historical overview of issues surrounding women as participants in biomedical research trials, followed by an ethical framework and discussion of the issues of informed consent, contraception requirements, and the appropriate inclusion of individuals in research studies. This document does not address research activities pertaining directly to fetal tissues, which are covered by the ACOG Statement in Support of Fetal Tissue Research. The issue of informed consent in clinical care is covered in more detail in Committee Opinion No. 819, Informed Consent and Shared Decision Making in Obstetrics and Gynecology.
ACOG recognizes and supports the gender diversity of all patients who seek obstetric and gynecologic care. In original portions of this document, the authors seek to use gender-inclusive language or gender-neutral language. When describing research findings, this document uses gender terminology reported by the investigators. ACOG's policy on inclusive language can be reviewed at https://acog.org/clinical-information/policy-and-position-statements/statements-of-policy/2022/inclusive-language.
The document also takes into consideration the effect of historical and legal restrictions on the use of certain terminology, as well as the effect of stigma. Therefore, it includes the use of terms such as “woman,” “women,” “patient,” and “individual” and refers to source materials using these terms. The following distinctions also exist for the purposes of this document: “All people who could become pregnant” refers to people with uteri; “all people who could have become pregnant” refers to women who have undergone hysterectomy or transgender men who have undergone gender-affirming care; and “all people who identify as women” refers to women and transgender women.
History of Exclusion From Research TrialsWomen, both pregnant and nonpregnant, have traditionally been excluded from research trials. The history of the limited inclusion of women is punctuated by early failures and significant adverse events. The example of thalidomide is discussed in detail later. In response, early bioethicists and other advocates launched an effort to protect people who could become pregnant, who could have become pregnant, who are lactating, and who identify as women from the hazards of research (2). In 1977, the Food and Drug Administration (FDA) excluded people “of childbearing potential” from early drug research, dramatically limiting participation in medical research (3).
By the 1990s, these trends began to reverse. Significant efforts by the NIH in the early 1990s led to a meaningful increase in the proportion of women participating in research trials (4). Since then, further progress has been made, encouraged by the FDA and the NIH, culminating in further strategic initiatives to include women in medical research (5). As of January 21, 2019, the federal government's Common Rule, which governs oversight of national human subjects research, no longer labeled pregnant people as "vulnerable." This classification had been a barrier to inclusive research in the past (6). Although this represents recent progress from a regulatory perspective, knowledge gaps persist given a continued lack of inclusion of people who identify as women, pregnant people, and people who are breastfeeding in research trials. Thus, continued emphasis on recruitment into research must be encouraged.
All people who could become pregnant, who could have become pregnant, who are lactating, or who identify as women should be presumed eligible for participation in research studies, and their recruitment and participation should be encouraged regardless of race, ethnicity, sexual orientation, gender identity, or socioeconomic status.Historically, arguments advanced to defend the exclusion of women cited possible harms to future children. Although the risk of harm should always be minimized, the possibility of risk to fetuses or future children is not in itself sufficient to justify the exclusion of people from research. Critically, ongoing exclusion of these individuals from research trials will only perpetuate the paucity of applicable data and the flawed practice of applying male-derived research results to care for people who could become pregnant, who could have become pregnant, who are lactating, or who identify as women. The paucity of data has implications for pregnant people and the health of fetuses or future children because, for example, so few medications are studied to any meaningful degree in pregnancy, making clinical decision making and risk stratification difficult. In doing so, the risk of harm is thus shifted to those who are pregnant and lactating because they may need medications and treatments without the benefit of evidence for their safety. Furthermore, exclusion of people who could become pregnant, who could have become pregnant, who are lactating, and who identify as women from research out of concern for risks to fetuses or future children may disproportionately emphasize that risk. Thus, fixation on a potential for reproduction and risks to fetuses or future children may counteract efforts to increase our knowledge base about non–pregnancy-related issues, reinforce sex and gender bias, and signal that theoretical risks to future children outweigh potential risks of nonparticipation.
ETHICAL ISSUES AND CONSIDERATIONS JusticeBecause of a history of systematic exclusion of women from research, in 1993, Congress directed that women were to be included in all federally funded clinical investigations unless inappropriate (1). Consequently, the NIH now requires that women and members of minoritized populations and their subpopulations be included in all NIH-funded research “unless a clear and compelling rationale and justification establishes to the satisfaction of the relevant institute/center director that inclusion is inappropriate with respect to the health of the participants or the purpose of the research” (4). This requirement appears to have had an effect on inclusivity in research, at least for NIH-sponsored studies. In the 2022 fiscal year, of the 10.8 million research participants in NIH-sponsored studies, people who could become pregnant, who could have become pregnant, and who identify as women numbered about 6 million (55.8%) (7). Similar data, however, are not available from non–NIH-funded research, for which collection of demographic information may not be standardized and can be limited.
Ethical principles mandate fair distribution of benefits and burdens of participation in research, underpinning the need for equitable inclusion in medical research of people who could become pregnant, who could have become pregnant, who are lactating, or who identify as women.Justice requires that people who could become pregnant, who could have become pregnant, who are lactating, or who identify as women be included in studies in sufficient numbers to determine whether their responses to treatment are different from others and whether treatment options derived from research are equivalently applicable to all individuals (8).
Although the ostensible concern expressed and driving the exclusion of people who can become pregnant is the risk to fetuses or potential future children, the effect is a lack of evidence and guidance for medical management for all people who are not traditionally included in research. This affects commonly excluded individuals throughout their life span. People who can become pregnant or are pregnant are not typically permitted to enroll in studies of novel treatments for complex or chronic medical conditions. In addition, excluding or removing those who could become pregnant from research prevents learning about the potential fertility benefits of controlling chronic disease. Part of the solution to such broad exclusionary approaches should be appropriate informed consent processes and deference to the autonomous decision-making ability of all individuals.
Informed ConsentAn independent review of the risks and benefits of research by an appropriate body is fundamental to the formulation of any research protocol (9). Ethical implementation of a research protocol thereafter requires an appropriate informed consent process that ensures autonomous decision making. Researchers have an obligation to disclose and discuss with prospective participants all material risks. In the case of people who can become pregnant or are pregnant, this includes all material risks to fetuses or potential future children (10). Disclosure should include risks that are likely to affect an individual's decision to participate or not to participate in the research. Participation in studies with greater than minimal risk requires careful consideration.
Imposing a heavier burden of consideration on people who could become pregnant, who could have become pregnant, who are lactating, or who identify as women ahead of participation in studies represents a structural barrier to equal inclusion and should be avoided. Respect for autonomous decision making by all people, whether they can become pregnant or not, avoids imposing unethical obstacles to research participation.Researchers should be cautioned to avoid the therapeutic misconception in decision making regarding the offer and participation in research. The “[t]herapeutic misconception exists when individuals do not understand that the defining purpose of clinical research is to produce generalizable knowledge, regardless of whether the subjects enrolled in the trial may potentially benefit from the intervention under study or from other aspects of the clinical trial.” (11). Although some people who could become pregnant, who could have become pregnant, who are lactating, or who identify as women may find benefit from participation in a clinical trial, this should not be the primary purpose or goal. When “a research subject fails to appreciate the distinction between the imperatives of clinical research and of ordinary treatment, and therefore inaccurately attributes therapeutic intent to research procedures,” then therapeutic misconception has occurred (12).
Because the process of informed consent cannot anticipate all conceivable risks, participants must be made aware during this process that should they develop unanticipated complications during the research, they should contact the researcher or a representative of the IRB immediately. Individuals should be informed of all available therapeutic options. When people who could become pregnant or are pregnant have been exposed to more than minimal risk in the conduct of research, they should be encouraged to participate in any available follow-up evaluations to assess the effects on themselves and on their future children.
Contraception Requirements and Research TrialsMany research trials, regardless of the nature of the study, the potential for harm in pregnancy, or the expressed reproductive wishes of participants, require all who could become pregnant to use at least one reliable form of contraception yet do not require the same of participants who create sperm and could participate in reproduction.
A contraception requirement for people who could become pregnant, who could have become pregnant, who are lactating, or who identify as women to participate in pharmaceutical research is paternalistic, discriminatory, and exclusionary. Rather, people may desire counseling about the risks of a potential pregnancy during participation, with the ultimate decision about contraception left to the individual.Not all people engage in sexual behavior that can lead to pregnancy. People should be trusted to take into consideration warnings given ahead of participation in research regarding teratogenicity, as well as potential failings of contraceptive methods. Excluding people who do not agree to adhere to strict contraceptive requirements violates individual autonomy and unnecessarily excludes people who can become pregnant from participation in research. Finally, contraception requirements are not routinely applied to men (or any people who can produce sperm); thus, these requirements are applied disparately to different genders and are discriminatory. Some medications under study could have effects on sperm that would then affect a potential pregnancy. If the true goal of past requirements regarding contraception is protection of a potential fetus or child, equitable application of this requirement would require the same of participants or intimate partners of participants who produce sperm. To avoid such inequity, all people, regardless of gender, should be educated about known and potential reproductive risks associated with participation in research, trusted to assess the risk of conception during participation for themselves, and govern their own behaviors accordingly.
In studies that pose more than minimal risk to a potential fetus, the informed consent process should include a detailed discussion of potential risks and benefits that therefore may also include a review of contraception options and their efficacy. Options for undesired pregnancy for the participant should also be reviewed because abortion access varies significantly by location, insurance status, and other socioeconomic factors. If participants elect to use a contraceptive method, they should be assisted in choosing a method, including abstinence, according to their needs and values (13). If a participant is unable to access contraception of choice, researchers should have a method in place to assist in securing access. Regardless of contraceptive use, if a pregnancy occurs during study participation, individuals should receive further counseling and informed consent discussions and have the opportunity to continue or decline to continue participating in the research study if they accept any additional risks.
Intimate Partner or Intended Nongestational Parent ConsentResearch protocols can raise questions about the potential effect of the research on the participants' intimate partners. Some investigators and IRBs require consent from both the research participant and any intimate partner(s) (14).
In general, requiring participation consent from an intimate partner or nongestational intended parent is not warranted, ethically justified, or legally required in research studies exclusively intended to benefit fetuses.Such consent is appropriate only if there is a risk of the partner's exposure to an investigational agent and exposure to that agent carries more than minimal risk; if data will be collected regarding an intimate partner's impression of the investigational agent; or if the inclusion or exclusion criteria directly relate to an intimate partner, such as if testing of a partner is required for enrollment in the trial (ie, semen analysis or testing for a sexually transmitted infection). If it is determined that none of the aforementioned conditions apply, intimate partner or nongestational intended parent consent is unwarranted and unethical. Requiring intimate partner consent imposes unnecessary barriers to participation, violates confidentiality and privacy, interferes with choices regarding reproductive options, interferes with a person's right to make independent decisions about their health, and may put an individual at risk in the setting of intimate partner violence (14).
Specific Considerations: People Who Are Pregnant as Participants in Research There is an ethical imperative to increase the inclusion of people who can become pregnant, who are pregnant, and who are lactating in research to improve maternal and fetal health.Prior barriers included an overemphasis on avoiding risk to fetuses or future children. The pursuit of zero fetal risk is not attainable and results in significant risks to maternal and fetal health outside the research setting.
Although there is concern that including pregnant people in the study of new drugs potentially could cause fetal harm, it is critical to recognize that both excluding pregnant people and those who are breastfeeding or lactating from research and overreacting to potential harm lead independently to harm. Historical experiences demonstrate this effect. When thalidomide was approved in Europe after studies in nonpregnant patients and subsequently used by pregnant women, many children were born with limb malformations. Had appropriate studies of thalidomide been conducted in pregnant women before its endorsement, these risks would have potentially been known, and fewer children would have been born with limb malformations (15). Notably, none of the early trials considered harms to fetuses that might accrue from thalidomide ingestion by potential fathers; more recent research has shown that thalidomide is present in semen (16). Men who take thalidomide for clinical purposes are now cautioned to use synthetic or latex condoms or to avoid sexual contact (even in the context of vasectomy) with a woman who is pregnant or could become pregnant, at least through the first trimester.
In contrast, the introduction of Bendectin (pyridoxine [vitamin B6]/doxylamine) into the U.S. market is an example of undue caution as a response to the history of thalidomide. In 1956, the combination of doxylamine, dicyclomine (an antispasmodic agent), and vitamin B6 was approved by the FDA and introduced into the U.S. market as Bendectin, an effective agent for the treatment of nausea and vomiting in pregnancy (17). In the years that followed, numerous lawsuits alleging association with birth defects were filed, ultimately resulting in the voluntary withdrawal of Bendectin from the market in 1983. This occurred despite the publication of research before and after the withdrawal of Bendectin that failed to detect evidence that the medication was linked to specific birth defects (18–21). A large meta-analysis including 170,000 exposures further demonstrated the doxylamine–pyridoxine combination to be safe in pregnancy and to cause no increase in adverse fetal effects (22). It was not until 2013, after ongoing proof of a lack of credible evidence of an association between Bendectin and birth defects, that the drug was reintroduced for clinical use (with FDA approval) for nausea and vomiting of pregnancy (23). In this case, premature and unfounded withdrawal of a medication for lack of pregnancy-specific investigation was followed by the development of a large body of appropriate pregnancy-specific research that ultimately disproved decades of concern.
One primary reason that pregnant people have been systematically excluded from research is their perceived status as “vulnerable.” The original language of the Common Rule included pregnant individuals and “children, prisoners, mentally disabled persons and economically or educationally disadvantaged persons” as a vulnerable population with respect to research participation because of concerns regarding barriers to appropriate informed consent in this population (6). ACOG, along with prominent organizations and scholars, advocated to remove pregnant people from this category because pregnancy does not impair autonomous decision making (24). In the revised Common Rule, the federal government removed the designation of pregnant people as a vulnerable population (6). Moving forward, further efforts on the local level of the IRB are needed to promote the inclusion of pregnant people in clinical trials (25).
It is important to note that research conducted during pregnancy is the only opportunity to study interventions aimed at conditions unique to pregnancy. Similarly, research during the process of labor and delivery is vital to improving care for pregnant people and their newborns. Pregnant people entering labor or in labor are still fully capable of consenting to participate in research.
A pregnant person in labor is able to undergo the appropriate informed consent process for research, similar to individuals with conditions that may have parallel connotations to labor, including other painful conditions or even those that are life-threatening or emergent (eg, appendicitis, nephrolithiasis, and myocardial infarction).A significant proportion of pregnant people undergo therapies aimed at managing nonobstetric medical conditions. Studies have estimated that more than 60% of pregnant women use at least one prescription medication during their pregnancies (26). Most of these medications have not been studied specifically in pregnancy. The risk status for pregnant people and fetuses is unknown for the vast majority of FDA-approved medications. Had these drugs been studied in pregnancy early in their use, data on risk may have provided an opportunity to better balance the risks and benefits of their use. Because pregnancies are increasingly occurring in older individuals and in those with complex medical conditions, the use of prescription medications by pregnant people is likewise increasing (27, 28). Physicians who care for pregnant people with complex medical conditions and the pregnant individuals themselves are faced with making health care decisions based on insufficient clinical evidence.
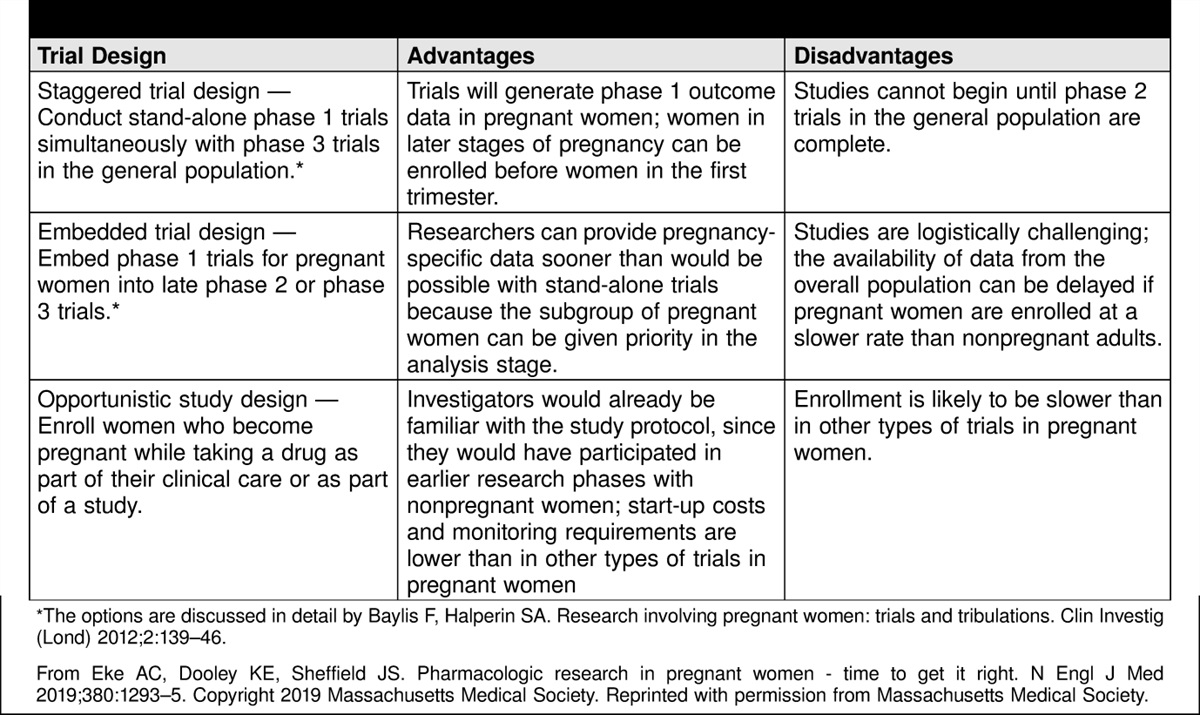
Research in pregnant people requires thoughtful study design. Baylis and Halperin outline specific study designs to feasibly include pregnant people in early drug studies (Table 1) (29). In 2009, the National Institute of Allergy and Infectious Diseases conducted a tiered research trial of the H1N1 vaccine (30). This study was conducted after initial safety and efficacy information for the general population was available, and it specifically evaluated pregnant people in the second and third trimesters to minimize inadvertent teratogenicity and to target the population most at risk of severe disease from H1N1 infection. Studies of this nature allow an incremental increase in risk as the chance of likely benefit increases.
 Table 1.:
Table 1.: Drug Trial Designs Suitable for Pregnant Women
The results of the H1N1 study involving pregnant people support the idea that it is possible to responsibly reform human research protection guidelines to enroll more pregnant individuals in research. This represents a shift from protecting populations from research to protecting populations through research.
Appropriate study design can maximize the benefits of enrolling people who can become pregnant, who are pregnant, and who are lactating while minimizing the risks to them, their fetuses, and their neonates. Special Considerations: The Fetus as the Focus of Research TrialsPotential treatments aimed specifically at the fetus also may be the focus of research trials. The overarching goal of fetal interventions is clear: to improve the health of children by intervening before birth to correct or treat prenatally diagnosed abnormalities (31). Any investigational fetal intervention, however, has implications for the pregnant person's health and body integrity and therefore cannot be performed without consideration of their well-being or without their explicit informed consent (31). It is impossible to enroll the fetus in a research trial without affecting the pregnant person either physically (in the case of surgical treatments or radiologic studies, including ultrasonography) or pharmacologically (as in the case of medications given to the pregnant person that then cross the placenta to treat the fetus). Because the pregnant person who chooses to undergo these research procedures and treatments must assume some of the risk, respect for their autonomy requires a thorough discussion of the risks and harms specific to the pregnant person (32). A pregnant person's right to informed refusal must be respected fully. The potential benefit to the fetus should not be overstated in an effort to secure a pregnant person's participation. Similarly, it is essential that undue risks to a person's health are not undertaken if the likely benefit to the fetus is minimal. Safeguards should be in place to protect people considering enrollment in fetal research studies (31). One possible safeguard would be to involve a research participant advocate with no direct ties to the experimental protocol who can act as an independent advocate for the pregnant person, especially when the proposed fetal intervention poses significant risks to the pregnant person (33). Such advocates should be nondirective in their support of the pregnant person's decision. Involving someone who understands the culture of research but maintains separation from the research team can provide an ethical safeguard to support the pregnant person (34).
Consent of the pregnant person alone is sufficient for most research. When research has a significant chance of harm to the fetus, consent of the father of the pregnancy or other nongestational parent may also be required by federal regulations (35) (Table 2). These regulations regarding nongestational parental consent have generated vigorous debate. They are not ethically justifiable and are an infringement on the pregnant person's autonomy. They may pose additional psychological or physical risks to the pregnant individual or their offspring. Furthermore, these requirements are not consistent with the wide variety of family structures that exist and may lead to the exclusion of potential study participants based on their family structure or the way they became pregnant.
 Table 2.:
Table 2.: Selected Federal Regulations on Informed Consent for Participants in Human Research*
ACOG supports a pregnant person's autonomy in making decisions. Deference to decision making by the nongestational parent during pregnancy infringes on and weakens autonomy of the pregnant person.
Generally, pregnant individuals' consent is sufficient for research interventions that affect them or their fetuses, as in other clinical scenarios. CONCLUSIONThe potential for pregnancy should not automatically exclude a person from participating in a study, although risks specific to pregnancy or potential pregnancy may need to be included in the informed consent process. Inclusion of people who could become pregnant, who could have become pregnant, who are lactating, or who identify as women in research studies is necessary for accurate data about health and disease. The generalization of results from trials conducted in men may yield erroneous conclusions that fail to account for biological differences. Although many improvements have occurred since the time of systematic exclusion of people who can become pregnant, who could have become pregnant, who are lactating, or who identify as women from research trials, more work needs to be done on the design of research trials so that they do not constrain the reproductive choices of study participants or unnecessarily exclude people who are pregnant or breastfeeding. Researchers and funding organizations should be aware of historical examples of exclusion and should strive for recruitment inclusivity. It is only by the inclusion of people who can become pregnant, could have become pregnant, are lactating, or identify as women in thoughtfully designed research studies that we can ultimately protect these populations and their offspring from potential risks of medications, treatments, or exposures.
CONFLICT OF INTEREST STATEMENTAll ACOG committee members and authors have submitted a conflict of interest disclosure statement related to this published product. Any potential conflicts have been considered and managed in accordance with ACOG's Conflict of Interest Disclosure Policy. The ACOG policies can be found on acog.org. For products jointly developed with other organizations, conflict of interest disclosures by representatives of the other organizations are addressed by those organizations. The American College of Obstetricians and Gynecologists has neither solicited nor accepted any commercial involvement in the development of the content of this published product.
REFERENCES 1. NIH Revitalization Act of 1993, Pub. L. No. 103-43. Accessed March 11, 2024. https://govinfo.gov/content/pkg/STATUTE-107/pdf/STATUTE-107-Pg122.pdf 2. Denny C, Grady C. Research involving women. In: Emanuel EJ, Grady C, Crouch RA, Lie RK, Miller FG, Wendler D, editors. The Oxford textbook of clinical research ethics. Oxford University Press; 2008. p. 407–22. 3. Office of Women's Health, U.S. Food and Drug Administration. OWH milestones and key events. Accessed February 27, 2024. https://fda.gov/consumers/owh-historical-information/owh-milestones-and-key-events 4. National Institutes of Health. NIH policy and guidelines on the inclusion of women and minorities as subjects in clinical research. Accessed February 27, 2024. https://grants.nih.gov/policy/inclusion/women-and-minorities/guidelines.htm 5. Office of Research on Women's Health. Advancing science for the health of women: the trans-NIH strategic plan for women's health research, 2019-2023. Accessed February 27, 2024. National Institutes of Health. https://orwh.od.nih.gov/sites/orwh/files/docs/ORWH_Strategic_Plan_2019_02_21_19_V2_508C.pdf 6. National Archives and Records Administration. Comparing the eCFR in effect on 7/19/2018 to what was previously in effect on 7/18/2018. 45 C.F.R. § 46.107 IRB membership. Accessed March 13, 2024. https://ecfr.gov/compare/2018-07-19/to/2018-07-18/title-45/subtitle-A/subchapter-A/part-46/subpart-A/section-46.107 7. Office of Research on Women’s Health. Report of the Advisory Committee on Research on Women's Health, fiscal years 2021–2022: Office of Research on Women's Health and NIH Support for Research on Women's Health. National Institutes of Health. Accessed February 27, 2024. https://orwh.od.nih.gov/sites/orwh/files/docs/ORWH_Biennial%20Report_121823_1516_F_508c_Optimized.pdf 8. Beauchamp TL, Childress JF. Principles of biomedical ethics. 8th ed. Oxford University Press; 2019. 9. National Bioethics Advisory Commission. Ethical and policy issues in research involving human participants. Accessed February 27, 2024. https://bioethicsarchive.georgetown.edu/nbac/human/overvol1.pdf 10. National Archives and Records Administration. General requirements for informed consent. 45 C.F.R. §46.116. Accessed March 11, 2024. https://ecfr.gov/current/title-45/subtitle-A/subchapter-A/part-46/subpart-A/section-46.116 11. Henderson GE, Churchill LR, Davis AM, Easter MM, Grady C, Joffe S, et al. Clinical trials and medical care: defining the therapeutic misconception. PLoS Med 2007;4:e324. doi: 10.1371/journal.pmed.0040324 12. Lidz CW, Appelbaum PS. The therapeutic misconception: problems and solutions. Med Care 2002;40:V55–63. doi: 10.1097/01.MLR.0000023956.25813.18 13. Anderson JR, Schonfeld TL, Kelso TK, Prentice ED. Women in early phase trials: an IRB's deliberations. IRB 2003;25:7–11. doi: 10.2307/3563818 14. Sullivan KA, Little M, Rosenberg NE, Mtande T, Zimba C, Jaffe E, et al. Women's views about a paternal consent requirement for biomedical research in pregnancy. J Empir Res Hum Res Ethics 2018;13:349–62. doi: 10.1177/1556264618783834 15. Kim JH, Scialli AR. Thalidomide: the tragedy of birth defects and the effective treatment of disease. Toxicol Sci 2011;122:1–6. doi: 10.1093/toxsci/kfr088 16. Teo SK, Harden JL, Burke AB, Noormohamed FH, Youle M, Johnson MA, et al. Thalidomide is distributed into human semen after oral dosing. Drug Metab Dispos 2001;29:1355–7. 17. Geiger CJ, Fahrenbach DM, Healey FJ. Bendectin in the treatment of nausea and vomiting in pregnancy. Obstet Gynecol 1959;14:688–90. 18. Mitchell AA, Rosenberg L, Shapiro S, Slone D. Birth defects related to Bendectin use in pregnancy, I: oral clefts and cardiac defects. JAMA 1981;245:2311–4. doi: 10.1001/jama.245.22.2311 19. Zierler S, Rothman KJ. Congenital heart disease in relation to maternal use of Bendectin and other drugs in early pregnancy. N Engl J Med 1985;313:347–52. doi: 10.1056/NEJM198508083130603 20. Holmes LB. Teratogen update: Bendectin. Teratology 1983;27:277–81. doi: 10.1002/tera.1420270216 21. Sanders J. From science to evidence: the testimony on causation in the Bendectin cases. Stanford Law Rev 1993;46:1–86. doi: 10.2307/1229235 22. McKeigue PM, Lamm SH, Linn S, Kutcher JS. Bendectin and birth defects, I: a meta-analysis of the epidemiologic studies. Teratology 1994;50:27–37. doi: 10.1002/tera.1420500105 23. Gee RE, Wood SF, Schubert KG. Women's health, pregnancy, and the U.S. Food and Drug Administration. Obstet Gynecol 2014;123:161–5. doi: 10.1097/AOG.0000000000000063 24. Blehar MC, Spong C, Grady C, Goldkind SF, Sahin L, Clayton JA. Enrolling pregnant women: issues in clinical research. Womens Health Issues 2013;23:e39–45. doi: 10.1016/j.whi.2012.10.003 25. Payne P. Including pregnant women in clinical research: practical guidance for institutional review boards. Ethics Hum Res 2019;41:35–40. doi: 10.1002/eahr.500036 26. Daw JR, Mintzes B, Law MR, Hanley GE, Morgan SG. Prescription drug use in pregnancy: a retrospective, population-based study in British Columbia, Canada (2001-2006). Clin Ther 2012;34:239–49.e2. doi: 10.1016/j.clinthera.2011.11.025 27. Mitchell AA, Gilboa SM, Werler MM, Kelley KE, Louik C, Hernández-Díaz S. Medication use during pregnancy, with particular focus on prescription drugs: 1976-2008. Am J Obstet Gynecol 2011;205:e1–8. doi: 10.1016/j.ajog.2011.02.029 28. Haas DM, Marsh DJ, Dang DT, Parker CB, Wing DA, Simhan HN, et al. Prescription and other medication use in pregnancy. Obstet Gynecol 2018;131:789–98. doi: 10.1097/AOG.0000000000002579 29. Eke AC, Dooley KE, Sheffield JS. Pharmacologic research in pregnant women - time to get it right [published erratum appears in N Engl J Med 2019;381:194]. N Engl J Med 2019;380:1293–5. doi: 10.1056/NEJMp1815325 30. Sperling RS, Engel SM, Wallenstein S, Kraus TA, Garrido J, Singh T, et al. Immunogenicity of trivalent inactivated influenza vaccination received during pregnancy or postpartum. Obstet Gynecol 2012;119:631–9. doi: 10.1097/AOG.0b013e318244ed20 31. Maternal–fetal intervention and fetal care centers. Committee Opinion No. 501. American College of Obstetricians and Gynecologists. Obstet Gynecol 2011;118:405–10. doi: 10.1097/AOG.0b013e31822c99af 32. Informed consent and shared decision making in obstetrics and gynecology. Committee Opinion No. 819. American College of Obstetricians and Gynecologists. Obstet Gynecol 2021;137:e34–41. doi: 10.1097/AOG.0000000000004247 33. Neill KM. Research subject advocate: a new protector of research participants. Account Res 2003;10:159–74. doi: 10.1080/714906094 34. Silber TJ. Human gene therapy, consent, and the realities of clinical research: is it time for a research subject advocate? Hum Gene Ther 2008;19:11–4. doi: 10.1089/hum.2007.1217 35. National Archives and Records Administration. Protection of human subjects. 45 C.F.R., part 46. Accessed March 11, 2024. https://ecfr.gov/current/title-45/subtitle-A/subchapter-A/part-46
Comments (0)