Bioinformatic analysis
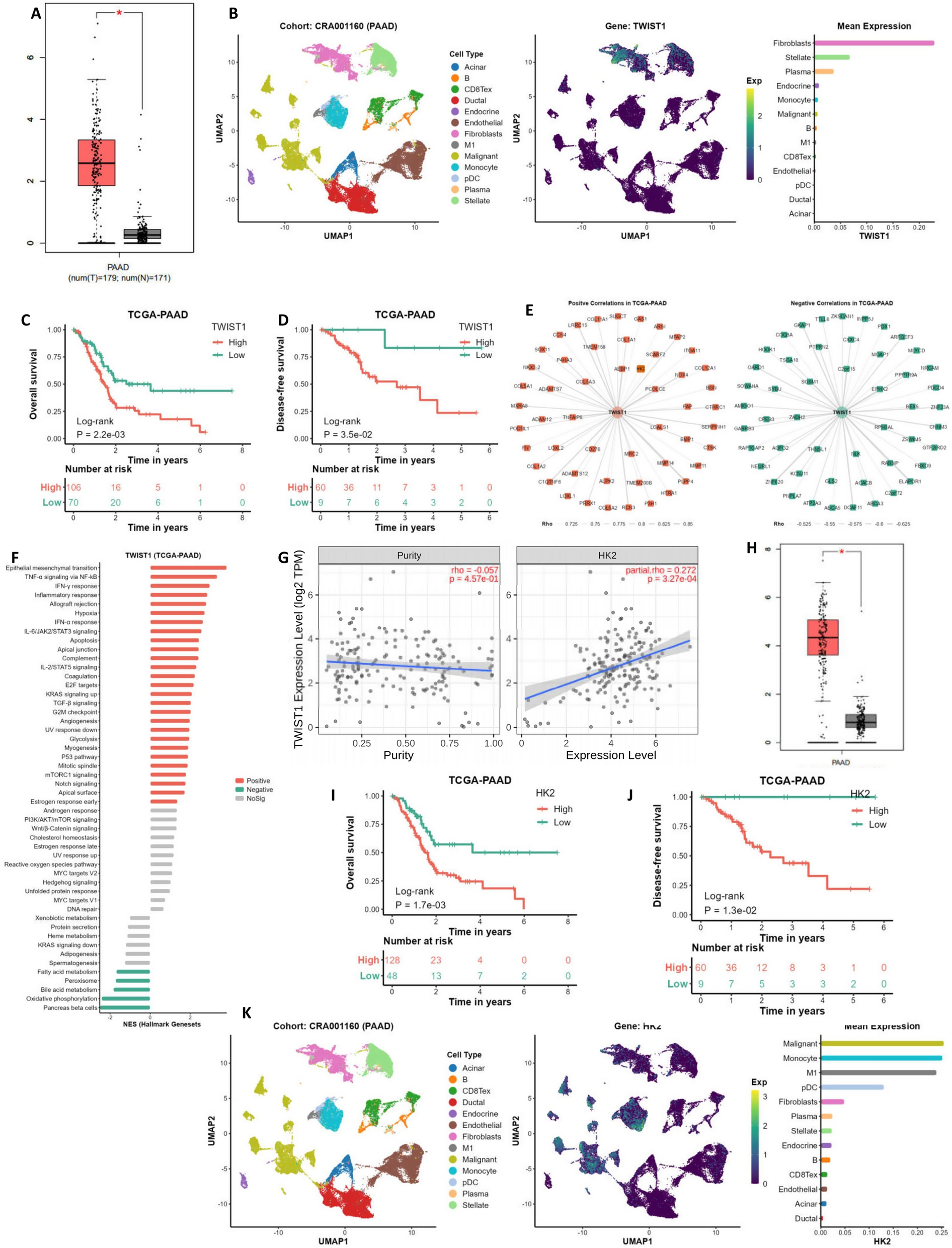
The gene expression and clinical data of pan-cancers and CESC (Cervical squamous cell carcinoma) were acquired from The Cancer Genome Atlas (TCGA) (https://tcga-data.nci.nih.gov/tcga/) and Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) databases (GSE45827, GSE29044 and GSE1456). These datasets were applied for the SLC25A17 expression and prognostic analysis. In SLC25A17 expression analysis, Willcoxon test was used to determine statistically significant differences between normal and tumor tissues. In Kaplan-Meier (K‐M) survival analysis, patients were divided into the low SLC25A17 group and high SLC25A17 group on the basis of the optimal cutoff value, which was determined using the surv_cutpoint function in the R survival package [12]. Samples in the TCGA dataset were divided into high SLC25A17 group (n = 835,76.4%) and low SLC25A17 group (n = 258, 23.6%). Samples in the GSE1456 dataset were divided into the high SLC25A17 group (n = 33, 20.8%) and low SLC25A17 group (n = 126,79.2%).
Cell culture and reagents
The human TNBC cell lines, namely MDA-MB-231, MDA-MB-468, and BT549, along with the normal mammary epithelial cell line MCF10A, were sourced from ATCC. MDA-MB-231 and MDA-MB-468 were grown in DMEM media containing 10% FBS. BT549 was grown in a specialized medium (CM-0041, Procell, China). MCF10A was cultured in a specialized medium (CL-0525, Procell, China). All cell lines were grown at a temperature of 37 °C in a humid environment with 5% CO2.
IL-6, chloroquine (CQ), N-acetylcysteine (NAC) and 3-methyladenine (3-MA) were purchased from MCE (Shanghai, China).
Cell transfection with lentivirus
The cells were plated in a 12-well plate with a cellular density of 1 × 105 cells per well. Once the cells adhered, lentiviruses (General Biol, Anhui, China) were mixed with Polybrene (5 mg/mL, General Biol) and added to the cells. Subsequently, stably transfected cells were selected by treating them with puromycin (Biosharp Life Sciences, Hefei, China) at a concentration of 2 µg/mL for a minimum of 2 weeks.
CCK8 and Colony forming cell assays
For CCK8 assay, 2 × 103 cells were planted into 96-well plates. After the specified incubation period, CCK-8 reagent (MedChemExpress, Shanghai, China) was added for an additional two hours. For colony forming analysis, TNBC cells were incubated for two to three weeks at a density of 500 cells per well in a six-well plate, followed by fixation in 4% paraformaldehyde and staining with 0.1% crystal violet.
EdU incorporation assay
EdU incorporation assays were carried out in accordance with the instruction of EdU Apollo®567 imaging kit (RiboBio, Guangzhou, China). 50 µM EdU solution was used to culture the cells for a period of 4 h. Subsequently, added 4% paraformaldehyde as a fixing solution and fix at room temperature for 25 min. Remove the fixative, add 0.3% Triton X-100 as a permeator, and incubate at room temperature for 10 min. Following that, the cells were incubated for 30 min using Apollo® staining solution, and the nuclei were stained using Hoechst 33342 solution.
Apoptosis and cell cycle analysis
The Annexin V-FITC/PI apoptosis detection kit (Yeasen, Shanghai, China) was used to carry out the apoptosis assay. Following their collection, TNBC cells were stained for 15 min at 25 °C using propidium iodide (PI) and Annexin V-fluorescein isothiocyanate (FITC). To perform the cell cycle assay, TNBC cells were gathered and underwent overnight fixation in 75% ethanol at a temperature of 4 °C. Subsequently, a staining process was carried out using the PI/RNase staining buffer for a duration of 15 min. The cell cycle and apoptosis assays were conducted using CytoFlex-LX flow Cytometer (Beckman, USA).
Wound healing and transwell assays
In wound healing examination, once the cells have reached complete fusion, the monolayer of cells is scraped using 200 µL pipette tips. The cells that had been scraped were washed with PBS. Subsequently, TNBC cells were cultured in a medium lacking of serum. At the designated time, a microscope was used to monitor the wound closure and photos were taken.
Transwell migration assays were carried out with 8.0 μm transwell chambers (Corning, NY, USA). To evaluate the cell invasion capacity, we utilized chambers filled with Matrigel (BD Science, MD, USA) in the upper chamber. The lower chamber was filled with 500 µL of culture media containing 20% FBS, while the upper chamber was filled with 100µL of cell suspension (5 × 104 cells/well). Following a designated incubation period, the cells in the upper chamber were removed, and 600 µL of 4% paraformaldehyde solution was added for fixation for 25 min. The fixing solution was then discarded, and 0.1% crystal violet solution was applied for staining for 25 min. Under a microscope, pictures of the invasion and migratory cells were captured.
Reactive oxygen species (ROS) assay
The levels of intracellular ROS were measured using Reactive Oxygen Species Assay Kit (S0033S, Beyotime, Shanghai, China). The cells were incubated with 10 mΜ DCFH-DA for 30 min at 37 °C in the absence of light, followed by three times washes. Subsequently, intracellular ROS production was measured by flow cytometry and fluorescence microscopy (Leica, MHG, Germany).
Monodansylcadaverine (MDC) staining
The autophagosomes were detected using MDC fluorescent staining kit (C3018S, Beyotime, Shanghai, China), following the manufacturer’s instructions. Cells were cultured in 24-well plates and treated with a 1:1000 dilution of MDC for 30 min at 37 °C in the absence of light. Then, the cells underwent three consecutive washes with assay buffer. The degree of autophagy was assessed utilizing a fluorescence microscope (Leica, MHG, Germany).
Transmission electron microscopy (TEM)
Cells were harvested and fixed using a commercially available electron microscope fixed solution (G1102, Servicebio, Wuhan, China) for 2 h at 4 °C. Subsequently, the cells were dehydrated using a series of graded ethanol concentrations (30%, 50%, 70%, 80%, 90%, 95%) and then infiltrated with propylene oxide before being embedded in an embedding medium overnight. Ultrathin sections were sliced with a Leica ultramicrotome and double-stained with uranyl acetate and lead citrate. Finally, images were acquired using a transmission electron microscope (Hitachi, Tokyo, Japan).
Autophagic flux analysis
The cells were seeded onto confocal dishes and subsequently infected with mRFP-GFP-LC3 adenoviral particles (HanBio, Shanghai, China). After 24 h, cells were fixed in 4% paraformaldehyde for 15 min, and the nuclei were stained using a DAPI solution. Images were obtained under confocal fluorescence microscope (Eclipse Ti-E, Nikon, Japan).
RNA extraction and real-time PCR (RT–PCR)
RNA was isolated utilizing Trizol method and subsequently reverse transcribed into complementary DNA. Subsequently, qRT-PCR analysis was carried out using the ChamQ™ SYBR® qPCR Master Mix (Vazyme, Nanjing, China) in accordance with the provided guidelines. The determination of mRNA expression was achieved using the 2−△△Ct approach. The primer sequences employed in this study are presented below: SLC25A17: F: 5'-GGTGGTAAACACCAGACTGAATNBC-3', R: 5'-ATNBCCGAGATTCCTTCATCTNBCGA-3'; GAPDH: F: 5'- GTCTCCTCTGACTTCAACATNBCG-3', R: 5'- ACCACCCTGTTTNBCTGTATNBCCAA - 3'.
Immunofluorescence staining
To perform immunofluorescence staining, the cultured cells were fixed using 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and then blocked with 3% BSA. Subsequently, the cells were incubated with antibodies against E-cadherin (1:200, 20874-1-AP, Proteintech), Vimentin (1:200, 60330-1-Ig, Proteintech), and LC3 (T55992, 1:200, Abmart) as well as the corresponding secondary antibody (Proteintech, 1:200, Wuhan, China). Afterward, the cells were stained with DAPI (Servicebio, Wuhan, China). Finally, the images were captured using a fluorescence microscope (Leica, MHG, Germany).
Western blotting analysis
Initially, the TNBC cells were lysed and the proteins were extracted using RIPA buffer (P0013B, Beyotime). Then, the proteins were separated through 10–15% SDS-PAGE and blotted onto 0.45 μm PVDF membranes (Millipore). Subsequently, the membranes were then underwent overnight incubation at 4 °C with primary antibodies as follows: anti-β-actin antibody (1:2000, 20536-1-AP, Proteintech), anti-SLC25A17 antibody (1:1000, A14840, Abclonal), anti-STAT3 (1:1000, #9139, CST), anti- Phospho-STAT3 (1:1000, #9145, CST), anti-JAK2 (1:1000, #3230, CST), anti-Phospho-JAK2 (1:1000, #3771, CST), anti-Ecadherin (1:20000, 20874-1-AP, Proteintech), anti-Vimentin (1:20000, 60330-1-Ig, Proteintech), anti-MMP2 (1:1000, 10373-2-AP, Proteintech), anti-MMP9 (1:1000, 10375-2-AP, Proteintech), anti- Cleaved PARP (1:1000, #5625, CST), anti-Cleaved Caspase-3 (1:1000, #9664, CST), anti-LC3 (1:1000, T55992, Abmart), anti-P62 (1:5000, T55546, Abmart), anti-Beclin-1 (1:1000, T55092, Abmart), anti-Bcl-2 (1:1000, T40056, Abmart), anti-Bax (1:1000, T40051, Abmart). Following primary antibody incubation, the membranes were exposed to secondary antibodies at room temperature for 1 h. Finally, the protein bands were visualized using the West Pico Plus Chemiluminescent Substrate (Thermo Fisher Scientific).
Immunohistochemistry (IHC)
Immunohistochemistry was conducted following the manufacturer’s instructions. In brief, slides were deparaffinized, rehydrated, subjected to staining using the primary antibodies against Ki67 (1:100, #9027, CST), Cleaved Caspase-3 (1:100, #9664, CST), Phospho-STAT3 (1:100, #9145, CST), LC3 (1:100, T55992, Abmart), SLC25A17 (1:100, A14840, Abclonal). The results were then evaluated by two professional pathologists independently.
Animal experiments
Female BALB/c nude mice (GemPharmatech, Nanjing, China) aged six weeks were employed to establish the xenograft model under specific pathogen-free (SPF) conditions. Each mouse received a subcutaneous inoculation of 100 µL of PBS containing 5 × 106 cells (MDA-MB-231 stably transfecting shNC and shSLC25A17) in the right axilla. Tumor volumes were measured every three days using digital calipers and calculated using the following formula: tumor volume (mm3) = 1/2 (length (mm) × (width (mm))2). After 24 days, the mice were sacrificed, and the tumor tissues were collected. All animal research was in compliance with the ARRIVE and was approved by the Animal Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology.
Statistical analysis
Data from all experiments were presented as mean ± standard deviation (SD) of three or more biological replicates. Statistical analyses were performed using GraphPad Prism 8, employing Student’s t-test for comparing two groups and one-way ANOVA for comparing more than two groups. P < 0.05 was considered statistically significant.





Comments (0)