
Remember me


Subarachnoid hemorrhage (SAH) is an acute catastrophic neurological disease and the third most common subtype of stroke. Nowadays the incidence of SAH has decreased over the past decades, possibly because of smoking cessation, management of hypertension and other lifestyle changes [1]. However, SAH still continues to be a serious health problem worldwide considering high morbidity and mortality, onset at a young age, and poor clinical outcomes. Secondary brain injury might be responsible for the poor outcomes of SAH, as secondary brain injury is longer and more serious compared with mechanical damage of first brain injury [2]. Currently, multiple physiological processes are responsible for secondary brain injury, such as toxically neuronal excitotoxicity, oxidative stress, and unresolved inflammation, which finally result in different types of cell death including apoptosis, necrosis, necroptosis, ferroptosis and so on [3]. Therefore, a strategy aiming to reduce secondary brain injury might interrupt the pathophysiological process of SAH and finally improve the outcomes of patients.
Ferroptosis is an iron-dependent form of programmed cell death [4]. The two main biochemical features of ferroptosis are iron accumulation and lipid peroxidation [5], which will result in the overwhelming accumulation of lethal lipid reactive oxygen species (ROS) and the abnormal activation of some lipid metabolism regulatory molecules [6]. Apparently, intracerebral hemorrhage followed by the lysis of red blood cells leads to the release of hemoglobin, which is the resource of iron overload and toxically induces ferroptosis. Many lines of evidence have demonstrated that inhibition of ferroptosis could attenuate brain injury in the model of SAH [7–9]. Moreover, dysregulation of ferroptosis contributes to several other diseases such as cancer, ischemia/reperfusion, and neurodegeneration (e.g. Alzheimer’s disease and Parkinson’s disease) [5,6], indicating that ferroptosis would be a promising therapy target.
Accordingly, a complex molecular biological network leading to lipid peroxidation in the process of ferroptosis has been intensively investigated, such as glutathione peroxidase 4 (GPX4) [10,11], FSP1-CoQ10 pathway [12], nuclear factor erythroid 2-related factor 2 (Nrf2) [13]. GPX4 is the key and best-defined molecular regulator in ferroptosis by reducing phospholipids and inhibiting lipoxygenase-mediated lipid peroxidation [10,11]. Recently, a new target named Peroxiredoxin6 (PRDX6) was proven to be involved in ferroptosis [14]. PRDX6 is a member of nonselenium peroxidases containing one cysteine that is different from PRDX1-5 and uses GSH as a physiological reductant to conduct peroxidase activities [15–17], which is similar to GPX4. Knockdown of intracellular PRDX6 significantly enhances ferroptotic cell death triggered by ferroptosis inducers (Erastin and RSL-3) via its phospholipase A2 (iPLA2) activity. In addition, knockdown of PRDX6 could not further enhance the level of GSH depletion related to ferroptosis, which is different from GPX4. However, the function and alteration of PRDX6 in SAH still lack of evidence. Considering that PRDX6 is an effective antioxidant protein, we are interested to know whether peroxidases PRDX6 is associated with ferroptosis in animal model of SAH.
Ferrostatin-1 (Fer-1) identified by Dixon et al. in 2012 is a small molecule inhibitor of ferroptosis and exhibits effective protection from neurological impairment in animal models of hemorrhagic stroke [8,18]. Thus, we repeated and confirmed the incidence of ferroptosis and the protection of Fer-1 from second brain injury in animal models of SAH. Then, we demonstrated that the protection of Fer-1 was associated with the upregulation of PRDX6. Moreover, the downregulation of PRDX6 in animal models of SAH could impair the neurological protection of Fer-1. Interestingly, the Fer-1 effect on the removal of MDA accumulation induced by SAH was impaired when PRDX6 was downregulated, but with no change in GSH deletion. In addition, the protection of Fer-1 targeting PRDX6 is dependent on its iPlA2 activity. Our in vivo results are consistent with previous in-vitro study showing that PRDX6 negatively regulates ferroptosis [14].
Methods AnimalsThe research protocols were approved by the Animal Care and Use Committee of Fujian Medical University and are in accordance with the guidelines of the National Institutes of Health. Adult male Sprague–Dawley rats (230–280 g) were purchased from the SLAC Laboratory Animal Co., Ltd. (Shanghai, China). The rats were kept in a 12-hour light/dark cycle and raised with free access to water and food under controlled temperature and humidity conditions.
Subarachnoid hemorrhage modelThe procedure to establish a rat model of SAH has been described in detail in a previous report [19]. Briefly, rats were anesthetized with an intraperitoneal injection of chloral hydrate (4 ml/kg) and kept in a supine position. The right common carotid artery and its bifurcations were separated. Then, a sharpened 4-0 monofilament nylon suture was inserted into the stump of the external carotid artery and advanced through the internal carotid artery until resistance was felt (length about 1.8–2.2 mm). The suture was pulled back slightly and advanced 3 mm further to puncture the bifurcation of the anterior and middle cerebral artery and then kept for 10–15 s. After that, the rats were returned to their cages to recover. Rectal temperature was maintained around 37 °C with a heating pad during the surgery. The sham-operated group underwent the same procedure without endovascular perforation. Rats were scored for neurological function 24 h after modeling and then euthanized to obtain brain tissue for subsequent experiments.
Drug administrationThe injection of Fer-1 and siRNA was carried out intracerebroventricularly (i.c.v.) before the induction of SAH as previously [20]. Fer-1 (Sigma-Aldrich, Missouri, USA) was dissolved in dimethyl sulfoxide (DMSO), PEG300, Tween80 and ddH2O to a final concentration of 25 mm and a single dose of Fer-1 (5 µl) was administered before the surgery. The detailed process was similar to that described by others (coordinates: 0.8 mm posterior, 1.5 mm lateral to bregma, 4 mm depth). The sham group received an equal volume of DMSO, PEG300, Tween80 and ddH2O in the same way. Commercially available in vivo siRNA of PRDX6 (si-PRDX6, sense primer 5-CUUCCACGAUUUCCUAGGATT-3 and antisense primer 5-UCCUAGGAAAUCGUGGAAGTT-3) were purchased, and designed from Riobio (Guangzhou, China), a solution containing 10 μl of si-PRDX6 (2μmol/L) was administered i.c.v followed by instruction. A specific inhibitor of Prdx6-iPLA2 activity 1-hexadecyl-3-(trifluoroethyl)-sn-glycerol-2 phosphomethanol (MJ33) was purchased from APEXBIO (Houston, Texas, USA) and administrated at a dosage of 0.5 μmol/kg, by tail vein 24 h before surgery of SAH induction.
Modified Perl’s iron stainingIron deposition in the paraffin sections of the brain was detected with the Prussian Blue Staining kit (Sbjbio Life Sciences, Nanjing, China). Briefly, sections were deparaffinized and hydrated to deionized water, then immersed in Prussian blue solution for 20 min, afterward the sections were washed with deionized water. Then, the sections were stained with an eosin staining solution and rinsed with deionized water again. Further, the sections were dehydrated, transparentized and mounted, and then section were imaged and evaluated the iron deposition in the cortex.
TEM analysisAfter 24 h of SAH, rats were perfused with PBS containing 1.5% paraformaldehyde and 3% glutaraldehyde. Basal cortical samples (1 × 1 × 1 mm3) were obtained and further fixed overnight at 4 °C in the same buffer. After fixation, tissue fragments were transferred to 1% osmium tetroxide and 1.5% potassium ferricyanide for 1.5 h. Then, the samples were dehydrated with different concentrations of ethanol and acetone and further embedded in epoxy resin and sectioned (80 nm). After staining with uranyl acetate and lead citrate, ultrathin sections were observed and photographed under a TEM.
Neurologic scoreModified Garcia score was used to assess neurologic performances 24 h after induction of SAH [19]. The modified Garcia scoring system mainly covers the following six indicators: (1) symmetry of limb movement; (2) forelimb extension activity; (3) spontaneous activity; (4) climbing ability; (5) trunk touch response; and (6) whisker touch response. The score for each indicator ranges from 0 to 3. Hence the maximum value is 18. In addition, a beam balance test was used to assess the exercise capacity of the rats by measuring the distance that the rats walked on the wood within 1 min. The score ranges from 0 to 4, and a higher score indicates the superior athletic ability.
SAH gradeThe severity of SAH was assessed with the grading system reported previously by two blinded researchers [8]. Briefly, rats were sacrificed 24 h post-SAH and perfused with ice-cold saline, then the brain tissues were rapidly removed and placed on ice. The basal surface of brain tissue was divided into six segments, and each segment was scored with a grade from 0 to 3 as follows: 0: no blood clot; 1: minimal blood clot; 2: moderate blood clot with recognizable arteries; 3: blood clot obliterating all arteries within the segment. The total score of the six segments ranged from 0 to 18, and revealed the bleeding scale of every SAH model.
Brain water contentThe severity of cerebral edema was commonly assessed by using the dry/wet weight method. Immediately after perfusion, the whole brain was removed. Each brain tissue was quickly weighed, and the procedure was triplicated to obtain the average value as the wet weight. We then place the brain tissue in a drying oven for a 24-h dehydration at 105 °C. The tissue was weighed to obtain the dry weight. The brain water content of rats could reasonably be calculated according to the formula below: brain water content = [(wet weight − dry weight)/(wet weight)] × 100%.
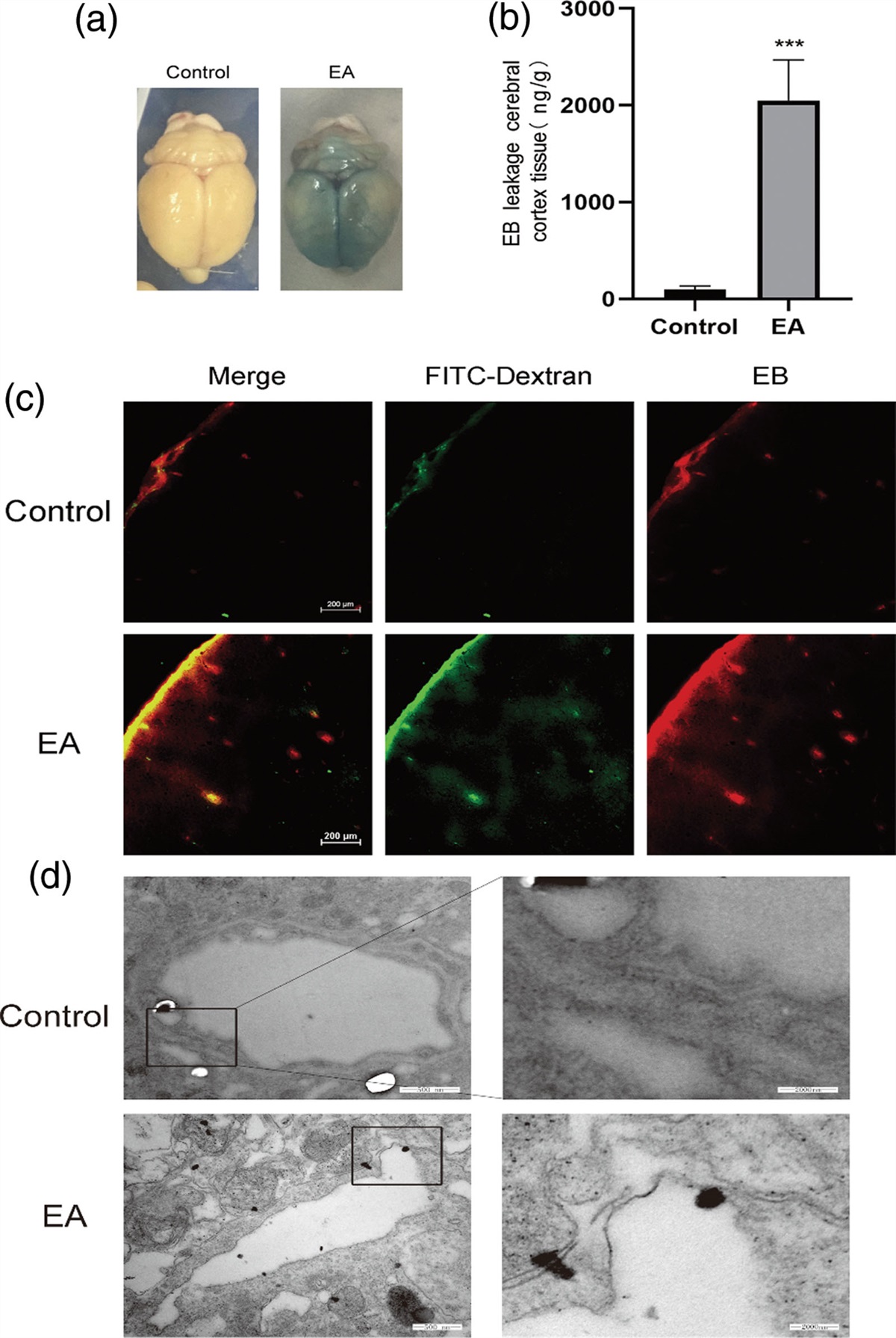
Evan’s blue leakage experimentBBB permeability was assessed 24 h after SAH as previously reported [8]. In brief, Evan’s blue dye (2%, 5 ml/kg; Sigma-Aldrich) was slowly injected into the right femoral vein and circulated for 1 h. After perfusion with ice-cold normal saline, brain samples were weighed, homogenized in 50% trichloroacetic acid (TCA), and centrifuged. The supernatant was mixed with ethanol and TCA, then incubated at 4 °C overnight. After centrifugation, the collected supernatant was spectrophotometrically quantified at 620 nm. Data are expressed as mean ± SEM after normalization with the sham group.
Determination of MDA, GSH, tissue iron and iPLA2 activityRats from each group were sacrificed after behavior tests, and the hippocampal and cortical tissues were separated and homogenized in homogenate liquid. Then, the mixture was centrifuged at 13 000 rpm for 10 min at 4 °C, and the supernatant was obtained for further biochemical analysis. The levels of MDA, GSH, tissue iron and iPLA2 activity were determined using commercial reagent kits based on manufacturers’ recommendations (Beyotime, Nanjing, China).
Western blotting analysisBrain tissue samples were collected at 24 h post-SAH, and the cortex on the basilar part, brain tissues were homogenized in ice-cold lysis buffer (Beyotime, Jiangsu, China). Total protein concentration was determined by the BCA protein assay (Beyotime). Equal amounts of protein were run on 12% SDS-polyacrylamide gel electrophoresis and then transferred onto PVDF membranes. After blocking with 5% milk in PBS for 1 h at room temperature, the membranes were incubated overnight at 4 °C with the following primary antibodies: PRDX6 (1 : 1000, Sigma, AV48268), β-Actin (1 : 4000; Sigma, A5441). Horseradish peroxidase-conjugated goat anti-rabbit (1 : 5000, Bioworld) was used as the secondary antibody the next day. Immunoreactivity was detected by enhanced chemiluminescence (Beyotime). Band intensities were quantified by using ImageJ software (http://rsb.info.nih.gov/ij/). The expression levels of proteins were expressed as the relative densities to β-Actin.
Quantitative real-time reverse transcription PCRTotal RNA was extracted using a rapid extraction kit (centrifugal column type; Bioteke, Jiangsu, China) and reverse transcribed into cDNA (2 μg total RNA per sample) using the High-Capacity cDNA Reverse Transcription Kit (Bioteke) according to the manufacturer’s instructions. The primer sequences used for the reaction were as follows: PRDX6 (forward primer, CCTGAAGAGGAAGCCAAACA, reverse primer, CAGTAGTATAGCGAGCGACCTA), β-actin (forward primer, CAACTGGGACGATATGGAGAAG, reverse primer, GGGACATGGCAGAAGAAAGA). The mRNA levels were standardized to β-actin, and the values were expressed as the foldchange of the threshold cycle value for the control by the 2−∆∆Ct method. Three independent experiments were performed.
Statistical analysisAll data are presented as mean ± SEM. Data were analyzed using the prism7.0 statistics software. One-way analysis of variance was performed followed by post-Turkey’s multiple comparisons test. A value of P < 0.05 was considered statistically significant.
Results Fer-1 inhibited ferroptosis in animal model of SAHFirstly, we intended to confirm evidence of ferroptosis in animal model of SAH. Representative images of brain were shown from control and SAH rats and TEM was employed to investigate mitochondrial morphologic changes. The membrane density of mitochondria was increased, and some mitochondria were shrunken and smaller in the SAH group, which are the typical mitochondrial morphologic changes of ferroptosis (Fig. 1a and b). As iron accumulation is one characteristic of ferroptosis, iron staining and quantification of tissue iron level were performed to investigating iron deposition. We found that iron deposition was significantly increased in SAH rats compared with control and sham groups, which could be inhibited significantly by Fer-1 (Fig. 1c and d).
 Fig. 1:
Fig. 1: Fer-1 inhibited the iron deposition in animal model of SAH. (a) Representative brain tissue and TEM images of rats in the control group, the area in the circle represents the sampling site for TEM and brain staining. (b) Representative brain tissue and TEM images of rats in the SAH group. (c) Iron staining image in each group, iron deposition indicated by red arrows. (d) Iron level in brain tissue, *P < 0.05, compared with Control. #P < 0.05, compared with SAH.
Fer-1 attenuated brain injury in the animal model of SAHCompared with the control group, SAH grades in the SAH groups were significantly increased, but Fer-1 treatment could not reduce subarachnoid bleeding significantly (Fig. 2a). Garcia JH scores were used to evaluate the neurological function, results showed that neurological scores were obviously lower after SAH, and treatment of Fer-1 significantly improved neurological disorder induced by SAH (Fig. 2b). Similar results in different brain anatomic parts were found in Evan’s blue leakage experiment and brain water content (Fig. 2c and d). As for Evan’s blue leakage experiment, higher BBB permeability was found in the SAH groups compared with control except for cerebellum, and Fer-1 administration could significantly improve the aggravation of EB extravasation (Fig. 2c). Brain water content was used to evaluate brain edema, and there were significant increases in bilateral hemispheres after SAH, while no changes in cerebellum and brainstem. Meanwhile, Fer-1 ameliorated the brain edema in the parts of the hemisphere (Fig. 2d).
 Fig. 2:
Fig. 2: Fer-1 attenuates brain injury in SAH. (a) SAH grading scores in each group. (b) The neurological scores assessed at 24 h after induction of SAH. (c) Evan’s blue leakage experiment at different anatomical structures used to determine BBB permeability. (d) Quantification of brain water content in each brain structure. *P < 0.05, compared with control, #P < 0.05, compared with SAH. BS, brainstem; C, cerebellum; LH, left hemisphere; RH, right hemisphere.
PRDX6 reduction and lipid peroxidation were alleviated by Fer-1 in animal model of SAHPRDX6 is an antioxidant peroxidase associated with oxidative stress and was found to be a negative regulator of ferroptosis currently [14]. However, the alteration of PRDX6 in animal model of SAH, and whether Fer-1 could affect its expression was still unknown. The protein and mRNA levels of PRDX6 determined by WB and RT-PCR respectively were both decreased after SAH, which were significantly alleviated by Fer-1 compared with negative control of DMSO (Fig. 3a and b). Regarding the important role of lipid peroxidation in ferroptosis and oxidative stress that is associated with secondary brain injury, we further used MDA and GSH as indicators of lipid peroxidation to investigate the regulation of Fer-1 in current research. Compared with the control group, the level of MDA was dramatically increased in the model of SAH (Fig. 3c), and GSH was reduced significantly (Fig. 3d), which were both counter-regulated by Fer-1 compared with negative control DMSO.
 Fig. 3:
Fig. 3: Fer-1 treatment reduces PRDX6 decline and lipid peroxidation in brain tissue after SAH. Protein and mRNA levels determined by western blot (a) and RT-PCR (b), respectively. MDA (c) and GSH (d) were quantified by commercial kits. *P < 0.05, compared with Control, #P < 0.05, compared with SAH.
Knockdown of PRDX6 impaired Fer-1 effect on lipid peroxidationAfter observing that the alteration of PRDX6 in SAH could be affected by Fer-1 treatment, we were interested to know whether PRDX6 was involved in the regulation of Fer-1 on lipid peroxidation. Therefore, commercial in vivo siRNA was employed to knockdown PRDX6 expression. As shown in Fig. 4a and b, although si-PRDX6 alone could not further decrease the expression of PRDX6 after induction of SAH significantly, the upregulation of PRDX6 protein and mRNA level induced by Fer-1 in SAH were significantly counteracted by si-PRDX6. Interestingly, the scavenging of MDA by Fer-1 was significantly antagonized by the administration of si-PRDX6 (Fig. 4c). However, knockdown of PRDX6 could not further dimmish the effect of Fer-1 on GSH deletion (Fig. 4d). Results were consistent with the previous study, PRDX6 knockdown could not further decrease GSH levels after Erastin treatment in H1299 cells and PRDX6 negatively regulates ferroptosis through its iPlA2 activity [14].
 Fig. 4:
Fig. 4: Knockdown of PRDX6 impaired Fer-1 effect on lipid peroxidation. (a) Protein level in brain tissue determined by western blot after SAH treatment with or without Fer-1 and si-PRDX6. (b) mRNA expression determined by RT-PCR in each group. Lipid peroxidation indicated by MDA (c) and GSH (d) evaluated by commercial kit in each group. *P < 0.05, compared with Control, #P < 0.05, compared with SAH, §P < 0.05, compared with SAH+Fer-1.
PRDX6 mediated the neuroprotection of Fer-1 from brain injury in SAHConsidering that lipid peroxidation is another character of ferroptosis, and knockdown of PRDX6 impaired the Fer-1 effect on lipid peroxidation, we further investigated whether si-PRDX6 could disrupt the neuroprotection of Fer-1. Neurological function indicated by Garcia JH scores in SAH+Fer-1+si-PRDX6 group returned to the level of SAH group compared with SAH+Fer-1 group (Fig. 5a). Similar results of BBB permeability and brain edema were observed. Compared with SAH+Fer-1 group, EB extravasation (Fig. 5b) and brain water content in the left hemisphere (Fig. 5c) significantly deteriorated again after administration of Fer-1 combined with si-PRDX6, which suggested PRDX6 mediated the neuroprotection of Fer-1 from brain injury in SAH.
 Fig. 5:
Fig. 5: Si-PRDX6 disrupted neuroprotection of Fer-1 in SAH. SAH grading scores (a), Evan’s blue leakage experiment (b), and brain water content in the left hemisphere (c) were evaluated as previously after administration with si-PRDX6. *P < 0.05, compared with Control, #P < 0.05, compared with SAH, §P < 0.05, compared with SAH+Fer-1.
Inhibition of iPLA2 activity suppressed protection of Fer-1 in SAHRegarding distinguishing effects of Fer-1 on MDA accumulation and GSH deletion in SAH, we further investigated whether iPlA2 activity of PRDX6 mediated Fer-1 therapeutic efficacy in SAH. Thus, a specific iPLA2 inhibitor (MJ33) was used to block iPLA2 activity in SAH, iPLA2 activity was determined first by commercial kit. As shown in Fig. 6a, SAH-induced dramatical decline of iPLA2 activity compared with control, which could be further inhibited by MJ33 significantly. In addition, treatment of Fer-1 could decrease the decline of iPLA2 activity after SAH, which may be because of the upregulation of PRDX6, as si-PRDX6 could further decrease iPLA2 activity significantly (Fig. 6a). Moreover, similar to si-PRDX6, although MJ33 alone could not induce greater MDA accumulation compared with SAH group, the removal of Fer-1 on MDA accumulation induced by SAH was inhibited by MJ33 (Fig. 6b). In the respect of GSH, whether MJ33 administration alone or combined with Fer-1 both had no effect on GSH deletion induced by SAH (Fig. 6c). After demonstrating that Fer-1 removed MDA through iPLA2 activity of PRDX6, we further investigated whether iPLA2 inhibitor MJ33 would affect the protection of Fer-1 from brain injury in SAH. Compared with SAH+Fer-1 group, neurological scores significantly decreased again after MJ33 treatment (Fig. 6d), and the improvement of BBB permeability deterioration indicated by EB extravasation by Fer-1 was also impaired by MJ33 (Fig. 6e), same as brain water content (Fig. 6f), which suggested that the protection of Fer-1 in SAH via iPLA2 activity of PRDX6.
 Fig. 6:
Fig. 6: IPLA2 activity was contributed to Fer-1 effects on MDA accumulation and brain injury. (a) IPLA2 activity determined by commercial kit after treatment of Fer-1 combined with or without MJ33, and combined with si-PRDX6. MDA level (b), and GSH (c) levels assessed in each group. neurological scores (d), Evan’s blue leakage experiment (e), and brain water content in the left hemisphere were evaluated to investigate brain injury in each group. *P < 0.05, compared with Control, #P < 0.05, compared with SAH, §P < 0.05, compared with SAH+Fer-1.
DiscussionIn the current study, we validate the neuroprotective effect of Fer-1 from brain injury in the early stage of SAH, demonstrated by improvements in neurological function, BBB permeability and brain edema in the animal model of SAH. Excessive accumulation of lipid peroxides was also significantly removed by the administration of Fer-1. Of importance, we provided evidence that the expression of a nonselenium peroxidase, PRDX6, reduced significantly after induction of SAH, and Fer-1 treatment could alleviate the reduction of PRDX6. Moreover, knockdown of PRDX6 by siRNA abolished the protection of Fer-1. All the above evidences suggest that PRDX6 may participate in the ferroptosis process of SAH. In addition, by using si-PRDX6 and iPLA2 activity inhibitor MJ33, lipid peroxides MDA removal by Fer-1 in animal model of SAH was impaired, however, the GSH level was not affected. Meanwhile, the neuroprotection of Fer-1 indicated by neurological function, BBB permeability and brain edema was also abolished by employing si-PRDX6 and MJ33. Therefore, the therapeutic effect of Fer-1 on brain injury in SAH might be mediated by iPLA2 activity of PRDX6.
Fer-1, a ferroptosis-specific inhibitor, inhibits ferroptosis in various pathologies and disease states, such as neurodegenerative diseases [21], and hemorrhagic stroke [8,9,18,19]. Several lines of evidence demonstrate the neuroprotection of Fer-1 in ICH or SAH. For example, Li et al. found that administration of Fer-1 protected the hemorrhagic brain, reduced lipid ROS production and attenuated the increased expression level of PTGS2 and its gene product cyclooxygenase-2 ex vivo and in vivo [18]. And the administration of Fer-1 upregulated Fpn and GPX4, decreased the iron content, then improved early brain injury after induction of SAH [8]. Our research confirms the neuroprotection of Fer-1 from brain injury in animal model of SAH, and the regulation of Fer-1 in lipid peroxidation.
Of note, ferroptosis is a ROS-dependent form of cell death characterized by iron accumulation and lipid peroxidation [5,22] The lipid peroxidation is a free radical-driven reaction that primarily affects polyunsaturated fatty acids in cell membranes, the product of which gradually increases during ferroptosis, from the initial lipid hydroperoxides (LOOHs) to the later production of malondialdehyde (MDA) and 4-hydroxynenoals (4-HNE) [5,6]. Disruption of the antioxidant systems is contributed to overproduction and accumulation of lipid peroxidation products. Currently, two main antioxidant system involved in ferroptosis are investigated comprehensively, one is GSH/GPX4 antioxidant system [23,24], and the other one is FSP1/CoQ10 antioxidant system [12]. GSH is a core antioxidant that is formed by condensation of Glu, Cys and Gly. Inhibiting the synthesis and utilization of GSH could induce ferroptosis [11]. Meanwhile, GPX4 continuously reduce phospholipid hydroperoxide (PLOOH) to the corresponding hydroxyl derivatives in the presence of GSH [25]. GSH depletion or GPX4 inactivation would induce ferroptosis by breaking down PLOOH reduction, which results in lipid peroxidation [10,11,24]. Another efficient endogenous antioxidant system is the ferroptosis suppressor protein 1 (FSP1)/coenzyme Q10 (CoQ10) pathway [12]. FSP1 uses the reducing equivalents of NADP [NAD(P)H] to reduce CoQ10 to ubiquinol, which acts as a lipophilic radical-trapping antioxidant to remove lipid peroxides from phospholipid bilayers [12,22]. In our study, we demonstrated that GSH deletion and MDA accumulation in SAH was improved by Fer-1treatment, the mechanism under which may be associated with both GSH/GPX4 and FSP1/CoQ10 antioxidant system. As Liu et al. demonstrated that GPX4 expression was upregulated after Fer-1 treatment [8], and Yuan et al. investigated that SIRT1activation and Fer-1 treatment could suppress SAH-induced ferroptosis by upregulating the expression of GPX4 and FSP1 [9].
Besides GSH/GPX4 and FSP1/CoQ10 antioxidant system, peroxidase PRDX6 was identified as a new negative regulator of ferroptosis recently [14]. LOOH accumulation and ferroptosis cell death triggered by ferroptosis inducers (Erastin and RSL-3) was significantly enhanced by knockdown PRDX6 [14]. PRDX6 is a multifunctional enzyme widely expressed at an abundant level in all tissues that possess peroxidase activity, calcium-independent phospholipase A2 (iPLA2), and lysophosphatidylcholine acyl transferase (LPCAT) activity [17]. For peroxidase activity, PRDX6 uses GSH as the physiological reductant similar to GPX4 [26]. Both the peroxidase and iPLA2/LPCAT activities play important roles in the repair of cell membrane peroxidation and cell survival [27,28]. However, there was no evidence of whether PRDX6 was associated with the ferroptosis process induced by SAH. Our study observed that PRDX6 expression was downregulated by induction of SAH, which could be alleviated by Fer-1 treatment. Meanwhile, PRDX6 knockdown impaired the neuroprotection of Fer-1 from brain injury in SAH. In addition, unlike other programmed cell death, there are rare prominent indicators of ferroptosis, so the products of lipid peroxidation sometimes are used to be markers of ferroptosis, such as LOOH and MDA [5]. In the current research, the removal of MDA accumulation by Fer-1 was impaired by PRDX6 siRNA. Taking together, PRDX6 is involved in the inhibition of ferroptosis by Fer-1 in animal model of SAH.
Moreover, in respect of ferroptosis, only iPLA2 activity of PRDX6 rather than peroxidase activity by using GSH as reductant participates in the regulation of lipid peroxidation, which is demonstrated by that GSH deletion induced by Erastin and RSL-3 was not affected by knockdown of PRDX6 [14]. Similar results were obtained in our study, there was no difference in GSH deletion between the Fer-1 treated group and Fer-1 combined with si-PRDX6 treated rats after SAH, which indicated that peroxidase activity of PRDX6 was not associated with Fer-1 regulation of lipid peroxidation. Further, iPLA2 inhibitor (MJ33) administration could impair the removal of MDA accumulation by Fer-1 and the neuroprotection of Fer-1 in our research.
In fact, previous studies have indicated contradictory data about the alterations and functions of PRDX6 in neurological diseases. For example, the expression level of PRDX6 in neurodegenerative diseases from postmortem patient brains to animal models to cultured cells can be increased, decreased, or even unchanged [29]. And in early stage of ischemia stroke, the expression level of PRDX6 can be upregulated [30], but in a rat model of contusion spinal cord injury, PRDX6 is downregulated by the TNF-α activation associated with an impaired motor function [31]. Indeed, Peroxiredoxin 6 (PRDX6) is an antioxidant protein and plays an important role in different neurological disorders. Knockdown of PRDX6 and inhibition of iPLA2 activity may impair the physiological antioxidant system. Thus, we hypothesize that the increase of PRDX6 in some situations may be the self-protection feedback to stimulus.
In conclusion, our results suggest that PRDX6 is involved in the pathophysiological process of SAH, and the neuroprotection of Fer-1from brain injury in SAH may be partially mediated by antioxidant protein PRDX6 besides GSH/GPX4 and FSP1/CoQ10 antioxidant system, mechanism of which might be the improvement of lipid peroxidation via its iPLA2 activity.
AcknowledgementsThis work was supported by the National Natural Science Foundation of China (No. 81701363) and Joint Funds for the Innovation of Science and Technology, Fujian province (No. 2020Y9111).
WHQ, ZY, YLH, LYX and KDZ designed the research; WHQ, ZY and ZMP performed the research and analyzed data; WHQ and ZY wrote the article. All authors contributed to the manuscript revision, read, and approved the submitted version.
Conflicts of interestThere are no conflicts of interest.
References 1. Claassen J, Park S. Spontaneous subarachnoid haemorrhage. Lancet 2022; 400:846–862. 2. Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol 2014; 10:44–58. 3. Chen J, Li M, Liu Z, Wang Y, Xiong K. Molecular mechanisms of neuronal death in brain injury after subarachnoid hemorrhage. Front Cell Neurosci 2022; 16:1025708. 4. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012; 149:1060–1072. 5. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res 2021; 31:107–125. 6. Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther 2021; 6:49. 7. Kuang H, Wang T, Liu L, Tang C, Li T, Liu M, et al. Treatment of early brain injury after subarachnoid hemorrhage in the rat model by inhibiting p53-induced ferroptosis. Neurosci Lett 2021; 762:136134. 8. Li Y, Liu Y, Wu P, Tian Y, Liu B, Wang J, et al. Inhibition of ferroptosis alleviates early brain injury after subarachnoid hemorrhage in vitro and in vivo via reduction of lipid peroxidation. Cell Mol Neurobiol 2021; 41:263–278. 9. Yuan B, Zhao XD, Shen JD, Chen SJ, Huang HY, Zhou XM, et al. Activation of SIRT1 alleviates ferroptosis in the early brain injury after subarachnoid hemorrhage. Oxid Med Cell Longev 2022; 2022:9069825. 10. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 2014; 16:1180–1191. 11. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014; 156:317–331. 12. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019; 575:688–692. 13. Lee C. Collaborative power of Nrf2 and PPARgamma activators against metabolic and drug-induced oxidative injury. Oxid Med Cell Longev 2017; 2017:1378175. 14. Lu B, Chen XB, Hong YC, Zhu H, He QJ, Yang B, et al. Identification of PRDX6 as a regulator of ferroptosis. Acta Pharmacol Sin 2019; 40:1334–1342. 15. Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med 2005; 38:1543–1552. 16. Rhee SG, Kang SW, Chang TS, Jeong W, Kim K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life 2001; 52:35–41. 17. Fisher AB, Vasquez-Medina JP, Dodia C, Sorokina EM, Tao JQ, Feinstein SI. Peroxiredoxin 6 phospholipid hydroperoxidase activity in the repair of peroxidized cell membranes. Redox Biol 2018; 14:41–46. 18. Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017; 2:e90777. 19. Liu Z, Zhou Z, Ai P, Zhang C, Chen J, Wang Y. Astragaloside IV attenuates ferroptosis after subarachnoid hemorrhage via Nrf2/HO-1 signaling pathway. Front Pharmacol 2022; 13:924826. 20. Li C, Wu Z, Xue H, Gao Q, Zhang Y, Wang C, et al. Ferroptosis contributes to hypoxic-ischemic brain injury in neonatal rats: Role of the SIRT1/Nrf2/GPx4 signaling pathway. CNS Neurosci Ther 2022; 28:2268–2280. 21. Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 2014; 136:4551–4556. 22. Pan F, Xu W, Ding J, Wang C. Elucidating the progress and impact of ferroptosis in hemorrhagic stroke. Front Cell Neurosci 2022; 16:1067570. 23. Li FJ, Long HZ, Zhou ZW, Luo HY, Xu SG, Gao LC. System X(c) (-)/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front Pharmacol 2022; 13:910292. 24. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med 2020; 152:175–185. 25. Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med 2003; 34:145–169.
Comments (0)