
Remember me




LCH is a clonal neoplastic proliferation of Langerhans cells expressing CD1a, langerin, and S100 protein and recurrent genetic alterations involving RAS/RAF/MEK/ERK pathway.
EpidemiologyLCH remains a rare condition, with an estimated incidence of 2 to 10 cases per million children aged 15 years or younger. There is an approximal equal distribution in male and female patients [4, 5]. Based on data from the Surveillance, Epidemiology, and End Results program for 2010 to 2016, the incidence of LCH was highest in the first year of life and then declined steadily with age. Beyond the age of 21, the rate of cases remains relatively stable across the adult age range [6].
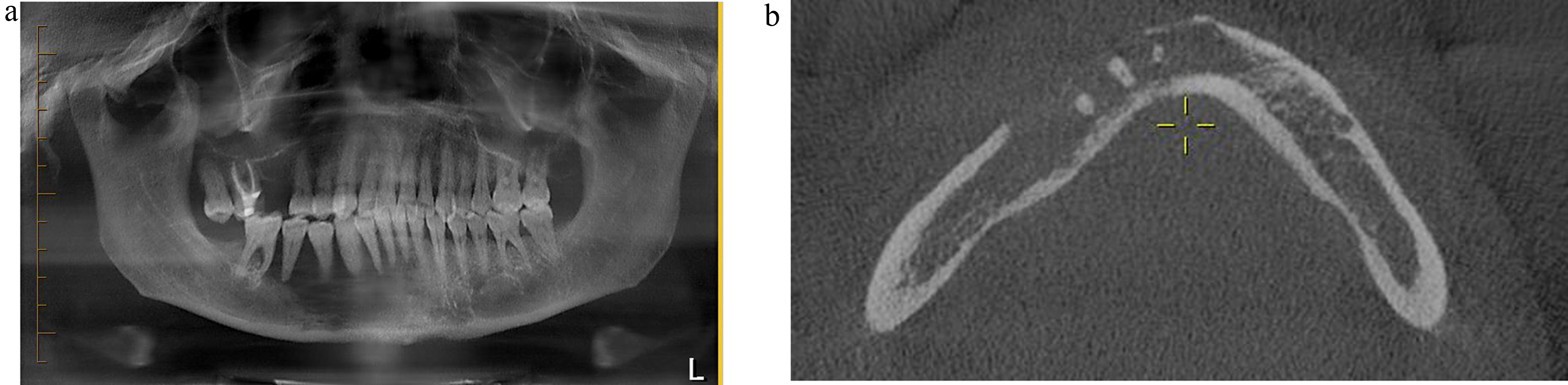
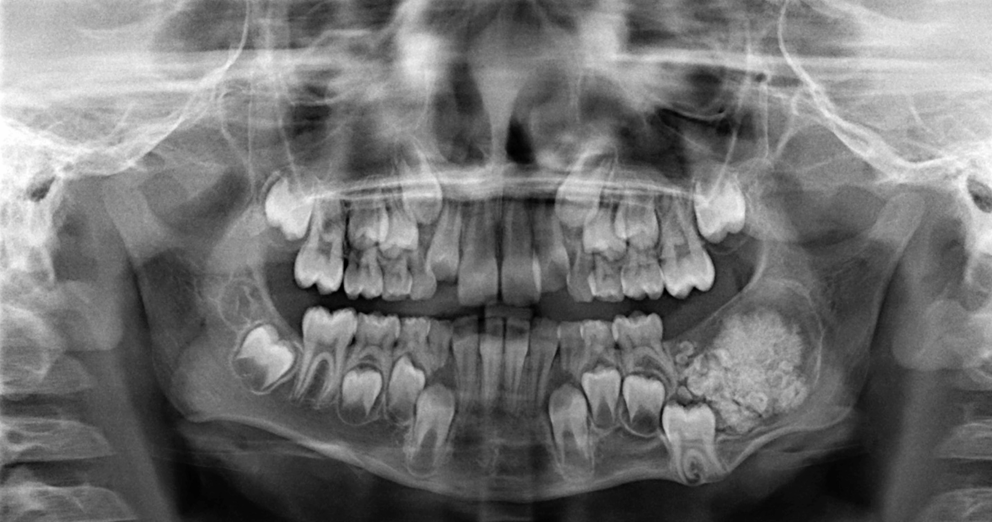
Clinical PresentationChildhood LCH varies clinically, from mild, single-organ involvement to severe, multisystem disease with symptoms ranging from asymptomatic bone lesions to life-threatening systemic illness [7, 8]. The skeleton is most affected, with up to 80% of cases presenting with bone lesions, which can appear radiographically as lytic (Fig. 1A) or widespread [9,10,11].
Fig. 1
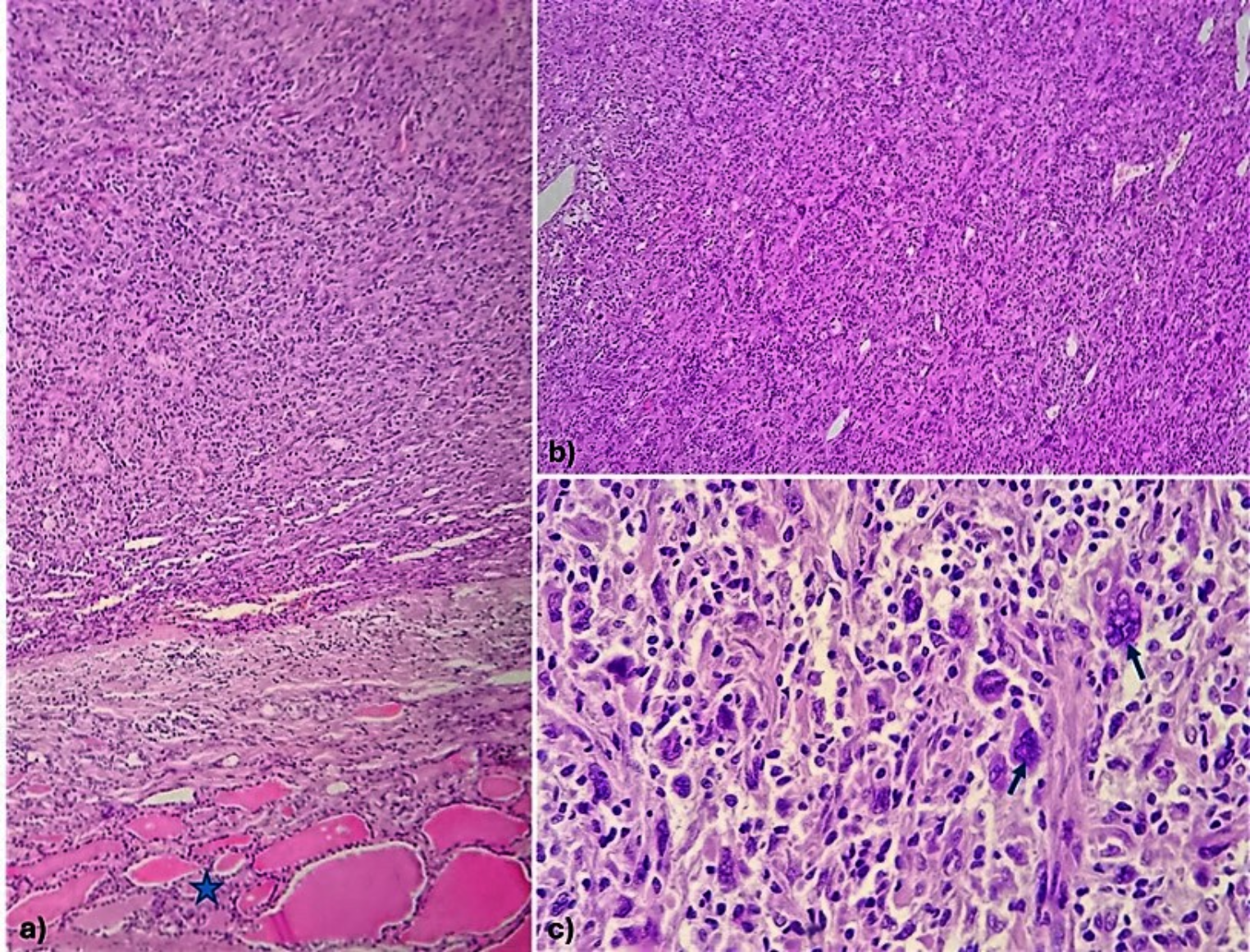
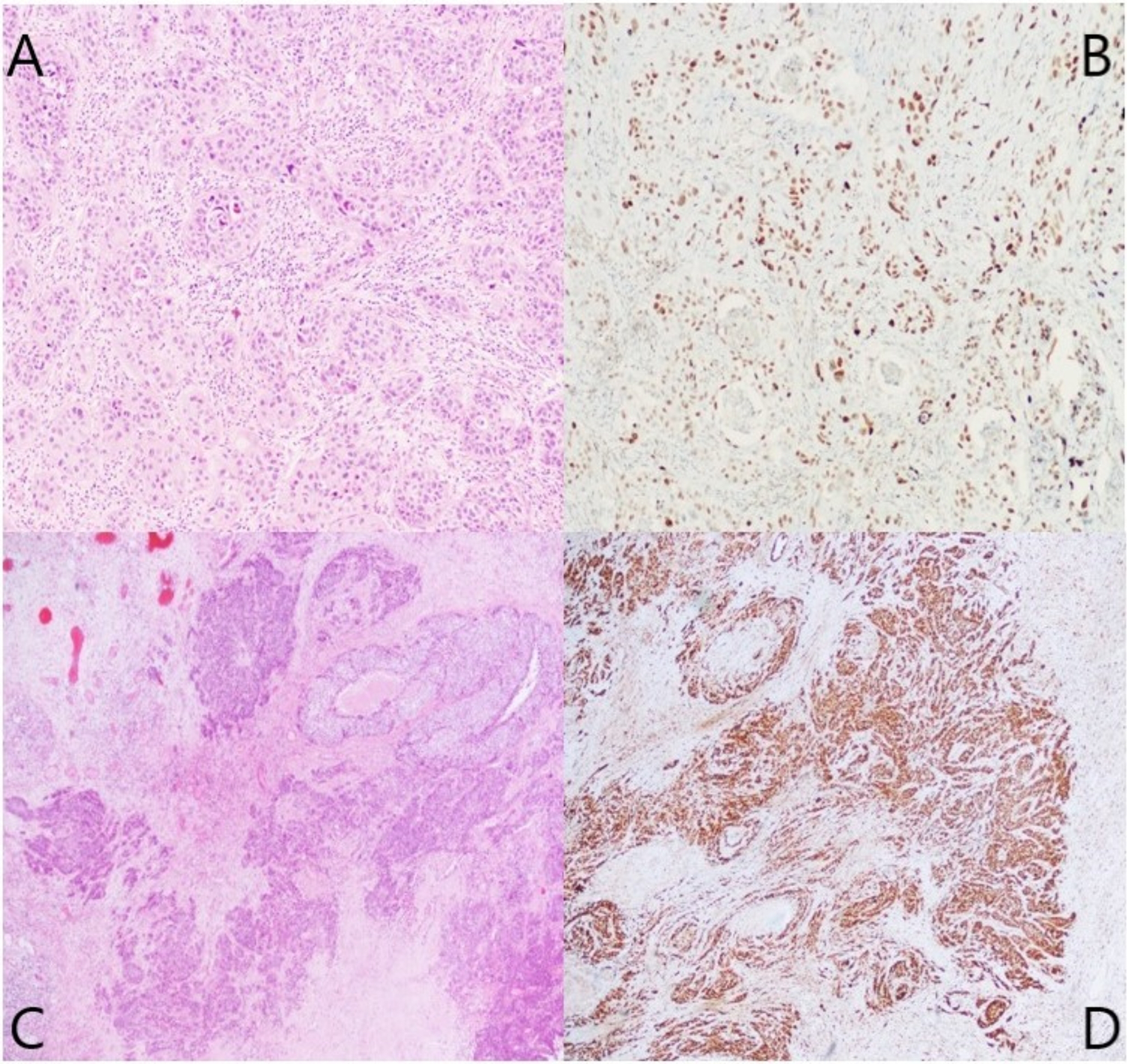
Langerhans cell histiocytosis. (A) A skull radiograph shows the typical appearance of lytic lesions of Langerhans Cell Histiocytosis in bone. The lesion exhibits scalloped edges and sharp borders with a radiodense focus that is commonly seen in skull lesions. Image from Wang G. Langerhans Cell Histiocytosis, in DP: Molecular Oncology, 3e, by Vasef M, et al. Copyright Elsevier 2024. Reused with permission. (B) The H&E section shows a sheet of oval-shaped Langerhans cells with eosinophilic cytoplasm, linear grooves, and inconspicuous nucleoli. Abundant eosinophils are present. (C) High-power view highlights the typical nuclear features of Langerhans cells. The nuclei are folded, show linear grooves, and often resemble coffee beans. (D) Immunostain CD1a diffusely marks Langerhans cells with positive membranous and cytoplasmic immunoreactivity. (E) Langerhans cells show a cytoplasmic granular staining pattern with langerin. (F) Langerhans cells show positive cytoplasmic and nuclear staining with S100
In adult LCH, most of the clinical and radiographic findings are like those of childhood LCH; however, isolated skin lesions are uncommon in adults. Radiographic bone lesions are present in 30–50% of cases, frequently involving the skull and dental sites, and less frequently pelvis, vertebrae, ribs, and extremities with the characteristic “punched-out” bony lesions [12]. Lung involvement in LCH can be part of multisystem disease or, more commonly in adults, can present as a primary or predominant form known as pulmonary LCH (PLCH). PLCH is a rare diffuse cystic lung disease, typically affecting young to middle-aged adults of both sexes and is seen almost exclusively in smokers or former smokers [13].
PathogenesisThe etiology of LCH has been a subject of significant research, evolving from theories of reactive processes to its current classification as a clonal neoplastic disorder. Langerhans cells are specialized, bone marrow-derived dendritic cells that act as antigen-presenting cells in the epidermis and mucosal tissues. They are uniquely identified by the markers CD1a, S100, and langerin (CD207) and contain distinctive Birbeck granules (“tennis racket” structures). Research suggests that circulating CD1c⁺ myeloid dendritic cells (mDCs) may serve as precursors to the pathogenic CD1a⁺CD207⁺ cells in LCH and, once activated via the ERK pathway, migrate from the bloodstream to lesion sites [14].
The neoplastic nature of LCH was first suggested by evidence of clonality in Langerhans cells through X-chromosome inactivation analysis. The presence of clonal histiocytes in all forms of LCH supports that it is a clonal neoplastic disorder rather than a reactive process with highly variable biological behavior [15].
The discovery of somatic mutations in the mitogen-activated protein kinase (MAPK) signaling pathway, especially BRAF V600E, has been key to understanding LCH pathogenesis. BRAF V600E, the most common mutation in LCH, occurs in 50–60% of cases and leads to continuous MAPK pathway activation, driving uncontrolled Langerhans cell proliferation [2]. In addition to BRAF, MAP2K1 mutations are found in 20–30% of LCH cases lacking BRAF mutations. Mutations in MAP2K1, which encodes the protein MEK1, lead to the constitutive activation of the pathway, promoting the uncontrolled proliferation of Langerhans cells—a hallmark of LCH [3, 16, 17]. MAP2K1 and BRAF mutations are mutually exclusive. Mutually exclusive somatic activating mutations in MAPK pathway genes have now been identified in approximately 85% of LCH lesions [18]. These mutations, more prevalent in aggressive forms of LCH, underscore the disease’s neoplastic nature and serve as targets for therapy [1].
While genetics is central to LCH pathogenesis, environmental and immune factors, such as viral infections (e.g., EBV) and exposures like tobacco smoke, are proposed contributors, though their precise roles remain unclear.
Immunological dysregulation significantly impacts LCH pathogenesis, with abnormal Langerhans cell proliferation accompanied by an inflammatory milieu of T cells, B cells, macrophages, and eosinophils, which suggests an immune response to the neoplastic cells. These immune cells, particularly T cells, secrete cytokines (e.g., IL-17, IL-6, and TNF-α) that contribute to the persistence of the lesions. The release of pro-inflammatory cytokines further promotes the abnormal proliferation of LCH cells and drives tissue destruction. Increased regulatory T cells (Tregs) and M2 macrophages in lesions further suppress the immune response, supporting LCH cell survival and lesion chronicity [19].
Eosinophils are a significant inflammatory component in LCH lesions, but their recruitment mechanisms and functional role in the disease remain poorly understood. An early study suggests that eosinophil recruitment may be mediated by CCL5 produced by Langerhans cells, but this finding has yet to be widely validated [20]. The precise contribution of eosinophils to LCH pathogenesis is not yet fully defined, underscoring the need for additional research to clarify their role in LCH.
Immune evasion is observed in LCH. Neoplastic LCH cells often express PD-L1, an immune checkpoint protein that interacts with PD-1 on T cells, leading to an inhibited immune response and allowing LCH cells to evade destruction [21]. This immune-tolerant environment supports the persistence of LCH cells and contributes to the chronicity of the disease.
DiagnosisDiagnosing LCH involves clinical assessment, histopathology, immunohistochemistry, molecular testing, and imaging to accurately identify the disease and assess its extent. A comprehensive evaluation of LCH has been outlined by Haupt et al. [22] Definitive diagnosis is based on pathology, with diagnostic Langerhans cells showing ovoid shape, grooved nuclei, fine chromatin, and minimal nuclear atypia (Fig. 1B-C); in some cases, the Langerhans cells have an increased Ki-67 proliferation index and mitotic activity. Along with Langerhans cells, a characteristic mixed inflammatory infiltrate is commonly seen, consisting of eosinophils, histiocytes, neutrophils, plasma cells, and small lymphocytes. Eosinophil-rich inflammatory infiltrates may also be seen in various conditions, such as allergic reactions, infections, and some malignancies. These add another layer of complexity to the diagnostic process and often require careful correlation with clinical, radiologic, and immunohistochemical findings to prevent misdiagnosis.
Langerhans cells stain positively for CD1a and langerin (Fig. 1D-E) as well as S100, showing both cytoplasmic and nuclear staining patterns (Fig. 1F), while markers like CD1c, B-cell markers, T-cell markers, CD30, and CD23 are negative. The BRAF V600E mutation-specific antibody (VE1) is positive in many cases, along with vimentin, CD4, and CD68, while CD163 positivity is only observed in 5–10% of cases. Immunohistochemistry for phosphorylated ERK (pERK) can also indicate MAPK pathway activity but is less specific than genetic testing [21]. On electron microscopy, Birbeck granules may appear but are not essential for diagnosis.
Molecular testing has dramatically advanced diagnosing and treating LCH by identifying genetic mutations in the MAPK pathway. Approximately 50–60% of LCH cases have the BRAF V600E mutation, which drives abnormal cell growth, while MAP2K1 mutations are found in an additional 20–30%, especially in BRAF-negative cases [21, 23]. Next-generation sequencing (NGS), Sanger sequencing, and pyrosequencing are standard methods for detecting these mutations. Use of these molecular tests significantly enhances the accurate diagnosis of this entity.
TreatmentRecent international expert consensus recommendations outline treatment options for adult LCH that vary based on disease extent, organ involvement, and genetic profile [12]. For patients with single-system LCH, surgery is often curative, with radiation as an alternative for inoperable lesions. Vinblastine and prednisone are standard first-line treatments for multisystem LCH, with more intensive chemotherapy reserved for high-risk organ involvement. Targeted therapies, including BRAF inhibitors (e.g., vemurafenib) for BRAF V600E mutations and MEK inhibitors (e.g., trametinib) for MAP2K1 mutations, show promise in refractory cases [21]. Supportive care, such as hormone replacement for pituitary involvement, is essential for quality of life.
PrognosisThe prognosis of LCH depends on disease extent, organ involvement, genetic mutations, and the responses to treatment. The prognosis in childhood LCH is guarded, as it is determined by the response to the initial therapeutic interventions, age of diagnosis, and extent of organ involvement. No response to initial therapy indicates a poor prognosis [24]. Unifocal LCH has a 5-year overall survival rate of over 90%. Multisystem LCH involving high-risk organs like the liver, spleen, or bone marrow has a more guarded prognosis, requiring systemic therapies such as chemotherapy or targeted treatments. Outcomes vary based on treatment response and specific organ involvement, with adult prognosis often affected by treatment efficacy and relapse frequency. Prognosis can range from no sequelae to severe organ dysfunction (e.g., liver or lung) or neurodegeneration. The presence of BRAF V600E mutations, often seen in aggressive LCH, correlates with higher relapse rates and multisystem disease. Targeted therapies, including BRAF and MEK inhibitors, have improved outcomes in high-risk patients and those unresponsive to standard treatments [25].
Comments (0)