
Remember me





Of the 473 patients prescreened at East Asian sites for FGFR2b overexpression, 45 were randomized to bemarituzumab plus mFOLFOX6 and 44 to placebo plus mFOLFOX6 (ITT; N = 89). The safety population included 44 patients in each treatment arm (ie, those who received at least one dose of the study treatment).
All patients had discontinued the study by the data cutoff date for the final analysis. Radiographic disease progression (n = 11 [24%] bemarituzumab plus mFOLFOX6, n = 25 [57%] placebo plus mFOLFOX6) and treatment-emergent adverse event (TEAE) (n = 21 [47%], n = 3 [7%], respectively) were the primary reasons for bemarituzumab/placebo treatment discontinuation. Death (n = 25 [57%] bemarituzumab plus mFOLFOX6, n = 32 [73%] placebo plus mFOLFOX6) was the primary reason for study discontinuation. Disease progression (n = 17 [38%] bemarituzumab plus mFOLFOX6, n = 27 [61%] placebo plus mFOLFOX6) was most common reason for death.
Baseline demographic and clinical characteristics were balanced between the treatment arms (Table 1). Across both treatment arms, the median age was 57 years and 63 (71%) were male. A total of 54 (61%) were enrolled in South Korea, 27 (30%) in China, 7 (8%) in Japan, and 1 (1%) in Taiwan. Gastric adenocarcinoma was the primary clinical diagnosis of cancer in 85 (96%) of the patients; ECOG status was 0 in 24 (27%) and 1 in 65 (73%); and 92% (n = 82) had disease stage IV at screening. One-third of patients received a single dose of mFOLFOX6 during screening (33% and 30% in the bemarituzumab plus mFOLFOX6 and placebo plus mFOLFOX6 arms, respectively). The median duration of exposure to study drug was 25.9 weeks (range, 2.0–94.6) in the bemarituzumab plus mFOLFOX6 arm and 27.4 weeks (range, 3.0–130.7) in the placebo plus mFOLFOX6 arm. The FGFR2b ≥ 10% subgroup comprised 29 (64%) patients from the bemarituzumab plus mFOLFOX6 arm and 31 (70%) patients from the placebo plus mFOLFOX6 arm.
Table 1 Patient demographics and characteristics in the FIGHT East Asian subgroupProgression-free survivalAt the final analysis data cutoff, median PFS follow-up time was 8.8 months (range, 0–35.9 months). PFS events occurred in 24 (53.3%) patients in the bemarituzumab plus mFOLFOX6 arm and in 32 (72.7%) patients in the placebo plus mFOLFOX6 arm. Median PFS was 12.9 months (95% CI 8.8–17.9 months) with bemarituzumab plus mFOLFOX6 and 8.2 months (95% CI 5.6–10.3 months) with placebo plus mFOLFOX6 (HR 0.50; 95% CI 0.29–0.87), with a 12 month estimated PFS rate of 55.5% (95% CI 37.5–70.2%) and 28.9% (95% CI 14.8–44.7%), respectively (Fig. 1a).
Fig. 1
Progression-free survival (a) and overall survival (b) in the FIGHT East Asian subgroup. Bema bemarituzumab plus mFOLFOX6, CI confidence interval, HR hazard ratio, mFOLFOX6 modified FOLFOX (infusional 5-fluorouracil, leucovorin, and oxaliplatin), mOS median overall survival, mPFS median progression-free survival, OS overall survival, Pbo placebo plus mFOLFOX6, PFS progression-free survival. HRs and 95% CIs were calculated using the unstratified Cox proportional hazards model. Vertical bars indicate censoring
Overall survivalAt the final analysis data cutoff, median OS follow-up time was 14.6 months (range, 0–40.5 months). A total of 25 (55.6%) patients in the bemarituzumab plus mFOLFOX6 arm and 32 (72.7%) patients in the placebo plus mFOLFOX6 arm died. Median OS was 24.7 months (95% CI 13.8–33.1 months) with bemarituzumab plus mFOLFOX6 and 12.9 months (95% CI 9.3–21.4 months) with placebo plus mFOLFOX6 (HR 0.56; 95% CI 0.32–0.96). The estimated OS rate at 12 months was 70.3% with bemarituzumab plus mFOLFOX6 and 59.5% with placebo plus mFOLFOX6; the rates at 24 months were 52.0% and 31.5%, respectively (Fig. 1b).
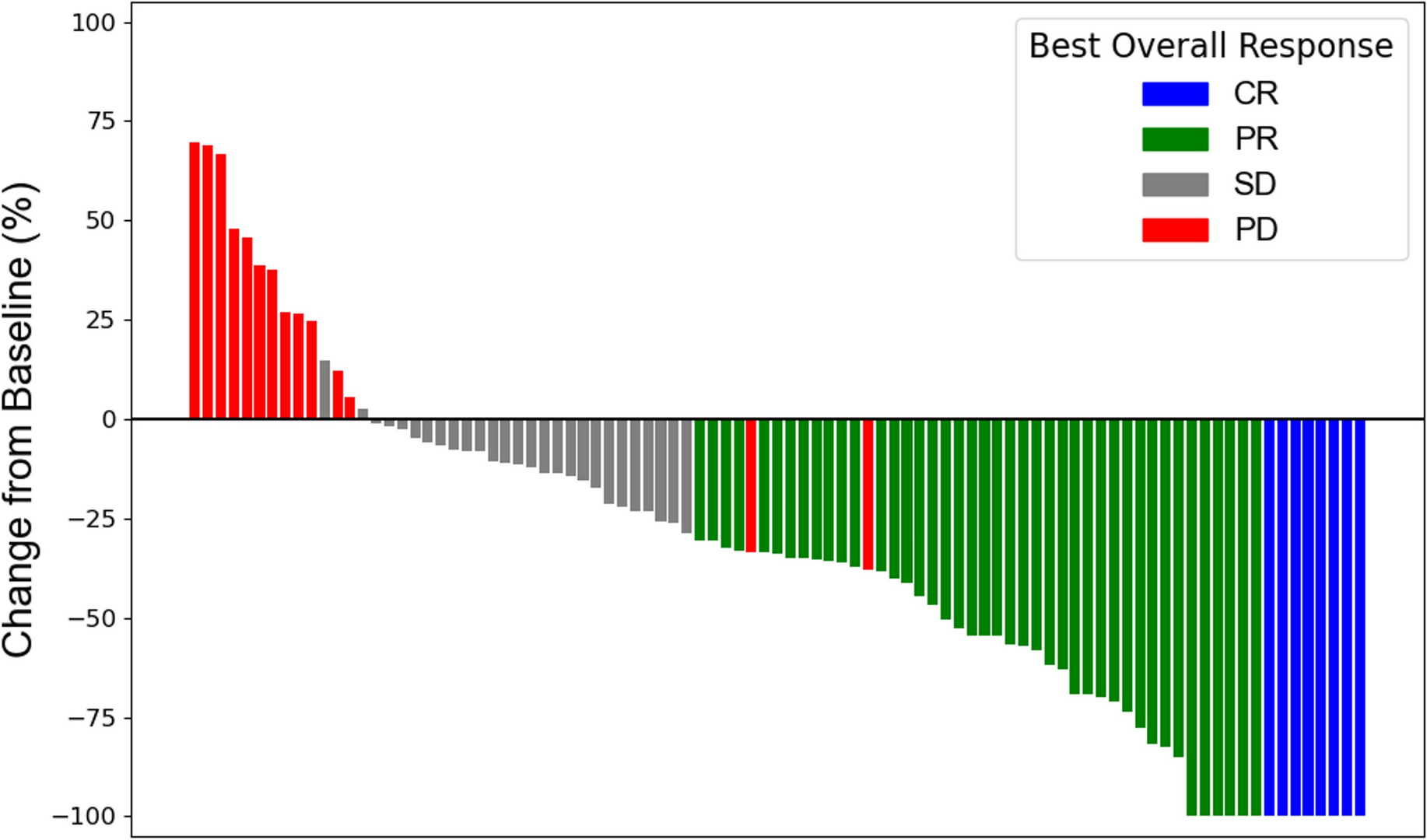
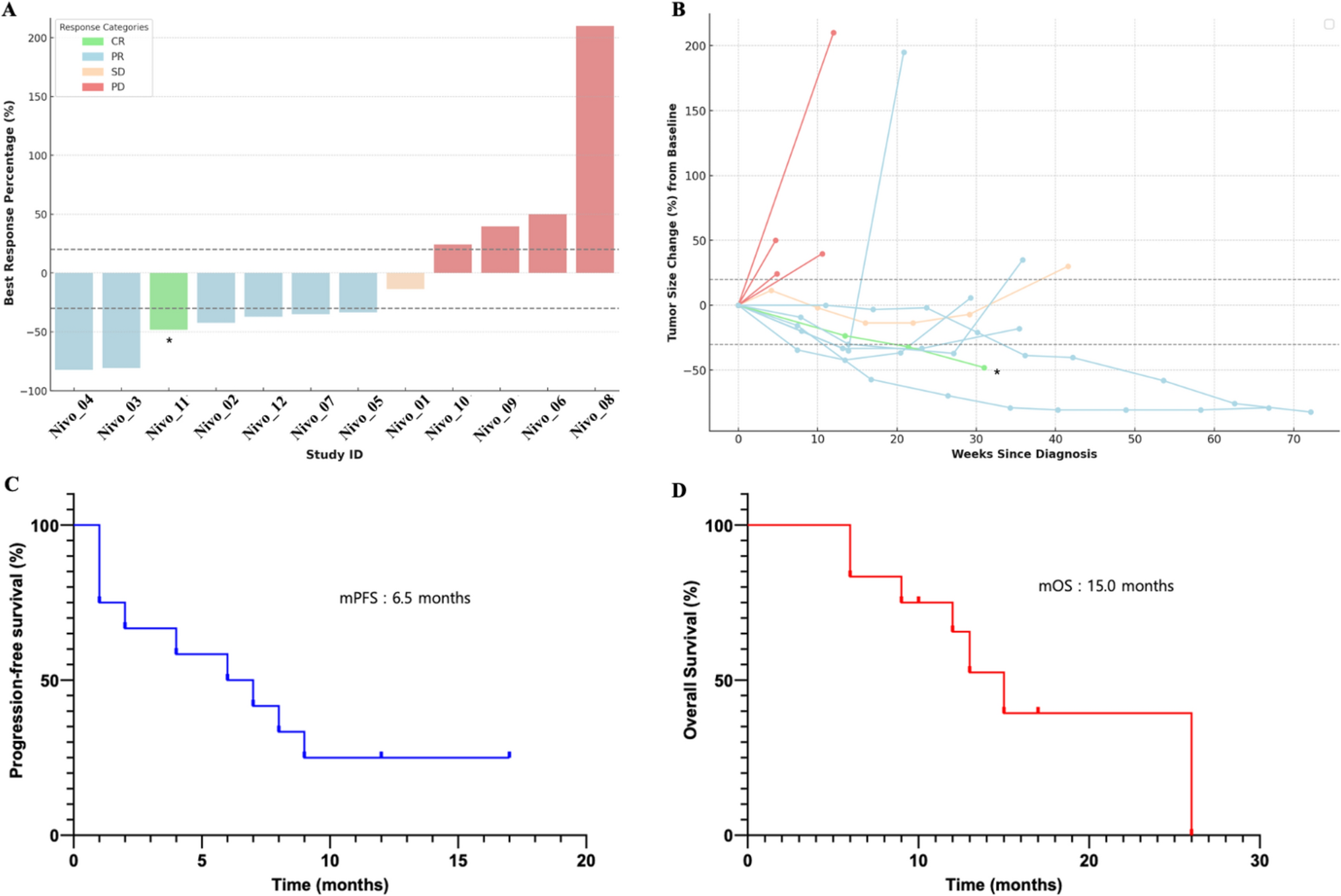
Response rateThe ORR was 48.9% (95% CI 33.7–64.2%) with bemarituzumab plus mFOLFOX6 and 34.1% (95% CI 20.5–49.9%) with placebo plus mFOLFOX6 (Table 2). CR and PR, respectively, was experienced by 3 (6.7%) and 19 (42.2%) patients in the bemarituzumab plus mFOLFOX6 arm and by 1 (2.3%) and 14 (31.8%) patients in the placebo plus mFOLFOX6 arm. Figure 2 displays the best percentage change in tumor size from baseline by individual patient. The median DOR was 15.6 months (95% CI 9.2–NR) with bemarituzumab plus mFOLFOX6 and 7.6 months (95% CI 3.7–14.8) with placebo plus mFOLFOX6.
Table 2 Tumor response in the FIGHT East Asian subgroupFig. 2
Response by patient in the FIGHT East Asian subgroup. Figure displays data from patients with measurable disease at baseline who were evaluable for evaluation of best overall response
Efficacy for patients in FGFR2b ≥ 10% subgroupEfficacy was assessed among patients in the FGFR2b ≥ 10% subgroup. Median PFS in this subgroup was 17.9 months (95% CI 9.5–NR) with bemarituzumab plus mFOLFOX6 and 7.6 months (95% CI 5.5–9.4) with placebo plus mFOLFOX6 (HR 0.28; 95% CI 0.13–0.57), with estimated 12 month PFS rates of 69.2% (95% CI 45.4–84.2%) and 24.1% (95% CI 9.9–41.6%), respectively (Supplementary Fig. 1a). Median OS was 30.1 months (95% CI 17.3-NR) with bemarituzumab plus mFOLFOX6 and 12.9 months (9.1–16.8) with placebo plus mFOLFOX6 (HR 0.43; 95% CI 0.22–0.86) (Supplementary Fig. 1b). The estimated OS rate at 12 months was 77.4% with bemarituzumab plus mFOLFOX6 and 58.6% with placebo plus mFOLFOX6; the rates at 24 months were 65.4% and 28.2%, respectively. The ORR was 51.7% (95% CI 32.5–70.6%) with bemarituzumab plus mFOLFOX6 and 35.5% (95% CI 19.2–54.6%) with placebo plus mFOLFOX6. CR and PR was experienced by 2 (6.9%) and 13 (44.8%) patients in the bemarituzumab plus mFOLFOX6 arm and by 0 and 11 (35.5%) patients in the placebo plus mFOLFOX6 arm.
Subsequent therapiesA total of 27 (61%) and 29 (66%) patients in the bemarituzumab plus mFOLFOX6 and placebo plus mFOLFOX6 arms, respectively, received second-line therapy after disease progression (safety analysis set) (Table 3). Taxanes (50%), pyrimidine analogues (27%), ramucirumab (26%), irinotecan (19%), and programmed cell death protein 1/programmed cell death ligand 1 (PD-1/PD-L1) inhibitors (15%) were the most common agents used as subsequent therapy.
Table 3 Subsequent anticancer therapy in the FIGHT East Asian subgroupSafetyAll patients in the bemarituzumab plus mFOLFOX6 arm and 43 (97.7%) in the placebo plus mFOLFOX6 arm reported TEAEs; 38 (86.4%) and 34 (77.3%) patients reported grade ≥ 3 TEAEs, respectively (Table 4). A total of 40 (90.9%) and 32 (72.7%) patients in the bemarituzumab plus mFOLFOX6 and placebo plus mFOLFOX6 arms, respectively, reported TEAEs related to bemarituzumab/placebo. A total of 21 (47.7%) and 4 (9.1%) patients in the bemarituzumab plus mFOLFOX6 and placebo plus mFOLFOX6 arms, respectively, reported TEAEs leading to discontinuation of bemarituzumab/placebo. Most TEAEs leading to discontinuation of bemarituzumab were corneal adverse events (17/21 [81%]); no corneal adverse events led to discontinuation of placebo. A total of 7 (15.9%) and 26 (59.1%) patients, respectively, in the bemarituzumab plus mFOLFOX6 arm and 7 (15.9%) and 22 (50.0%) patients in the placebo plus mFOLFOX6 arm reported TEAEs leading to dose reduction and dose delay of bemarituzumab/placebo.
Table 4 Incidence of treatment-emergent adverse events in the FIGHT East Asian subgroupA total of 14 (31.8%) and 18 (40.9%) patients in the bemarituzumab plus mFOLFOX6 and placebo plus mFOLFOX6 arms, respectively, reported serious TEAEs. A total of 3 (6.8%) and 2 (4.5%) patients in the bemarituzumab plus mFOLFOX6 and placebo plus mFOLFOX6 arms, respectively, reported fatal TEAEs.
A total of 14 (31.8%) patients in the bemarituzumab plus mFOLFOX6 arm and 0 patients in the placebo plus mFOLFOX6 arm reported grade ≥ 3 corneal adverse events; no patients reported serious or grade ≥ 4 corneal adverse events. Corneal adverse events, regardless of grade, were reported by 30 (68.2%) and 6 (13.6%) patients in the bemarituzumab plus mFOLFOX6 and placebo plus mFOLFOX6 arms, respectively; the median time to onset was 17.1 weeks (range, 0.1–38.6) and 12.4 weeks (range, 6.0–29.0), respectively (Table 5). A total of 24 (54.5%) and 1 (2.3%) patient(s) in the bemarituzumab plus mFOLFOX6 and placebo plus mFOLFOX6 arms, respectively, reported grade ≥ 2 corneal adverse events. In the bemarituzumab plus mFOLFOX6 arm, the median time to onset of grade ≥ 2 corneal adverse events was 28.6 weeks (range, 0.1–56.6). A total of 14 patients in the bemarituzumab plus mFOLFOX6 arm experienced resolution or downgrading of grade ≥ 2 corneal adverse events to grade 1, with a median time to resolution/downgrading of 24.5 weeks (range, 2.1–49.7). One patient in the placebo plus mFOLFOX6 arm experienced resolution of a corneal adverse event, with a time to resolution/downgrading of 2 weeks. All corneal adverse events resolved in 15 (34.1%) and 2 (4.5%) patients in the bemarituzumab plus mFOLFOX6 and placebo plus mFOLFOX6 arms, respectively.
Table 5 Summary of corneal adverse events
Comments (0)